Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Alignement de séquences biologiques
Objectifs poursuivis Alignement de séquences: généralités Alignement de deux séquences Recherche rapide de similarités dans les banques de séquences Alignement de n séquences (alignement multiple) Recherche de motifs dans les séquences
 Recherche de motifs dans les séquences.")
2
Objectifs poursuivis Les alignements permettent de comparer des séquences biologiques. Cette comparaison est nécessaire dans différents types d’études : Identification de gènes homologues Recherche de contraintes fonctionnelles communes à un ensemble de gènes ou de protéines. Prédiction de structure (ARN, protéine) Prédiction de fonction Étude des processus créateurs de variabilité entre les séquences. Reconstitution des relations évolutives entre séquences. Choix d'amorces PCR Construction de contigs (séquençage) ...
 Prédiction de fonction. Étude des processus créateurs de variabilité entre les séquences. Reconstitution des relations évolutives entre séquences. Choix d amorces PCR. Construction de contigs (séquençage) ...")
3
Analyse comparative des gènes de b-actine de l'homme et de la carpe

4
Prédiction de structure d'ARN

5
Phylogénie moléculaire

6
Alignement: représentation
Les résidus (nucléotides, acides-aminés) sont superposés de façon à maximiser la similarité entre les séquences. G T T A A G G C G – G G A A A G T T – – – G C G A G G A C A * * * * * * * * * * Mutations : Substitution (mismatch) Insertion Délétion Insertions ou délétions : indels (gap).
 sont superposés de façon à maximiser la similarité entre les séquences. G T T A A G G C G – G G A A A. G T T – – – G C G A G G A C A. * * * * * * * * * * Mutations : Substitution (mismatch) Insertion. Délétion. Insertions ou délétions : indels (gap).")
7
Quel est le bon alignement ?
G T T A C G A G T T A C G A G T T - G G A G T T G - G A * * * * * * * * * * OU G T T A C - G A G T T - - G G A * * * * * Pour le biologiste, généralement, le bon alignement est celui qui représente le scénario évolutif le plus probable Autres choix possibles (exemple: erreurs de séquençage pour la construction de contigs)
")
8
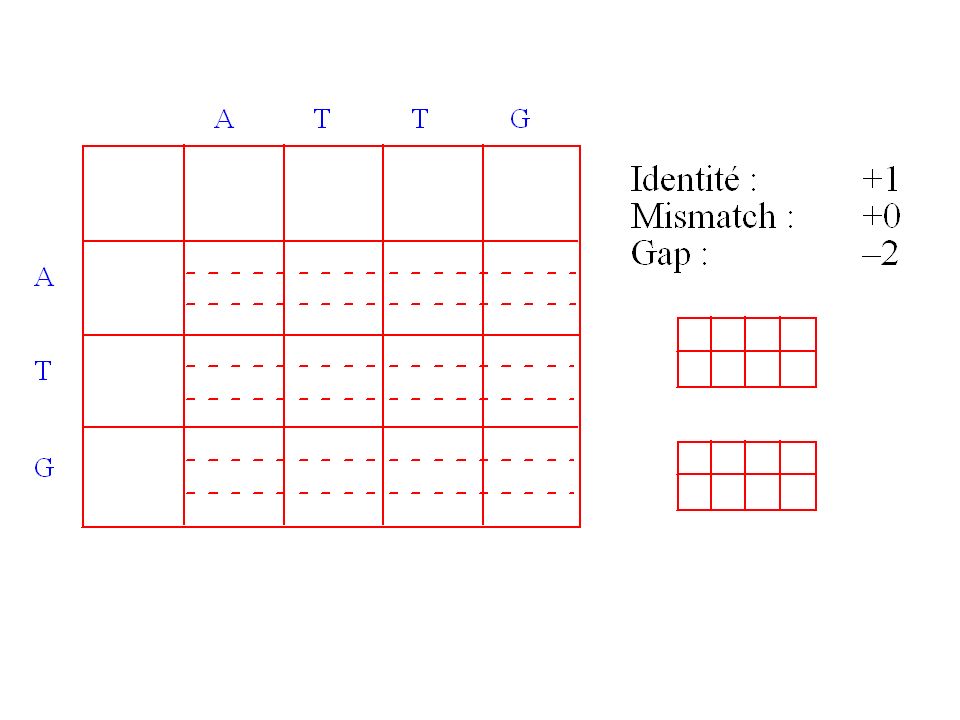
Fonction de score de similarité
G T T A A G G C G – G G A A A G T T – – – G C G A G G A C A * * * * * * * * * * Score = Exemple: identité = 1 mismatch = 0 gap = -1 Score = = 6

9
Modèle d'évolution (ADN)
Transition: A <-> G T <-> C Transversions : autres substitutions p(transition) > p(transversion) G T T A C G A G T T A C G A G T T - G G A G T T G - G A * * * * * * * * . * *
 > p(transversion) G T T A C G A G T T A C G A. G T T - G G A G T T G - G A. * * * * * * * * . * *")
10
Matrice de substitution (ADN)
Gap = -1 G T T A C G A G T T A C G A G T T - G G A G T T G - G A score = 4 score = 4.5

11
Modèle d'évolution (protéines)
Code génétique Asp (GAC, GAU) Tyr (UAC, UAU) : 1 mutation Asp (GAC, GAU) Cys (UGC, UGU) : 2 mutations Asp (GAC, GAU) Trp (UGG) : 3 mutations Propriétés physico-chimiques des acides-aminés (acidité, hydrophobicité, encombrement stérique, etc.) Matrices de Dayhoff (PAM), BLOSUM: mesures des fréquences de substitutions dans des alignements de protéines homologues PAM 60, PAM 120, PAM 250 (extrapolations à partir de PAM 15) BLOSUM 80, BLOSUM 62, BLOSUM 40 (basé sur des alignements de blocs) Substitutions conservatrices
 Tyr (UAC, UAU) : 1 mutation. Asp (GAC, GAU) Cys (UGC, UGU) : 2 mutations. Asp (GAC, GAU) Trp (UGG) : 3 mutations. Propriétés physico-chimiques des acides-aminés (acidité, hydrophobicité, encombrement stérique, etc.) Matrices de Dayhoff (PAM), BLOSUM: mesures des fréquences de substitutions dans des alignements de protéines homologues. PAM 60, PAM 120, PAM 250 (extrapolations à partir de PAM 15) BLOSUM 80, BLOSUM 62, BLOSUM 40 (basé sur des alignements de blocs) Substitutions conservatrices.")
12
Matrice de substitution (protéines)
D (Asp) E(Glu) F(Phe) G(Gly) W(Trp) M R D W - G F M R - D W G F M R - W D G F M R W D - G F * * * * * * * * * * Substitutions multiples (exemple: D E D)
 E(Glu) F(Phe) G(Gly) W(Trp) M R D W - G F M R - D W G F. M R - W D G F M R W D - G F. * * * * * * * * * * Substitutions multiples (exemple: D E D)")
13
Pondération des gaps TGATATCGCCA TGATATCGCCA TGAT---TCCA TGAT-T--CCA
**** *** **** * *** Gap de longueur k: Pénalités linéaires: w = do + de k do : pénalité pour l'ouverture d'un gap de : pénalité pour l'extension d'un gap

14
Pondération des gaps (plus réaliste)
Estimation des paramètres sur des alignements "vrais" (par exemple basés sur l'alignement de structures connues) Gap de longueur k: Pénalités logarithmiques: w = do + de log(k) w = f(log(k), log(PAM), résidus, structure) PAM: la probabilité d'un gap augmente avec la distance évolutive Résidus, structure: la probabilité d'un gap est plus forte dans une boucle (hydrophile) que dans le cœur hydrophobe des protéines
 Gap de longueur k: Pénalités logarithmiques: w = do + de log(k) w = f(log(k), log(PAM), résidus, structure) PAM: la probabilité d un gap augmente avec la distance évolutive. Résidus, structure: la probabilité d un gap est plus forte dans une boucle (hydrophile) que dans le cœur hydrophobe des protéines.")
15
Similarité globale, locale

16
Similarité, distance, homologie
Deux séquences sont homologues ssi elles ont un ancêtre commun 30% d'identité entre deux protéines => homologie, sauf si Fragment similaire court (< 100 aa) Biais compositionnel (régions de faible complexité, par exemple riche en Pro, Ala)
 Biais compositionnel (régions de faible complexité, par exemple riche en Pro, Ala)")
17
Le nombre d'alignements
Waterman (1984) a donné la formule récursive permettant de calculer le nombre total d’alignements possibles entre deux séquences comportant m et n résidus : D’autre part, Laquer (1978) a démontré que : Le nombre total d’alignements possibles entre deux séquences de même longueur croît de façon exponentielle.
 a donné la formule récursive permettant de calculer le nombre total d’alignements possibles entre deux séquences comportant m et n résidus : D’autre part, Laquer (1978) a démontré que : Le nombre total d’alignements possibles entre deux séquences de même longueur croît de façon exponentielle.")
18
Algorithmes d'alignement de deux séquences
Algorithme: description d'une suite d'opérations pour atteindre un objectif Calculer l'ensemble de tous les alignements possibles et garder celui de meilleur score Trop long (nombre d'alignements = f(exp(L)) Pas efficace (on recalcule souvent les mêmes valeurs) G T T A C G A G T T A C G A G T T - G G A G T T G - G A * * * * * * Algorithme de programmation dynamique Calcul de proche en proche de l'alignement optimal
) Pas efficace (on recalcule souvent les mêmes valeurs) G T T A C G A G T T A C G A. G T T - G G A G T T G - G A. * * * * * * Algorithme de programmation dynamique. Calcul de proche en proche de l alignement optimal.")
19
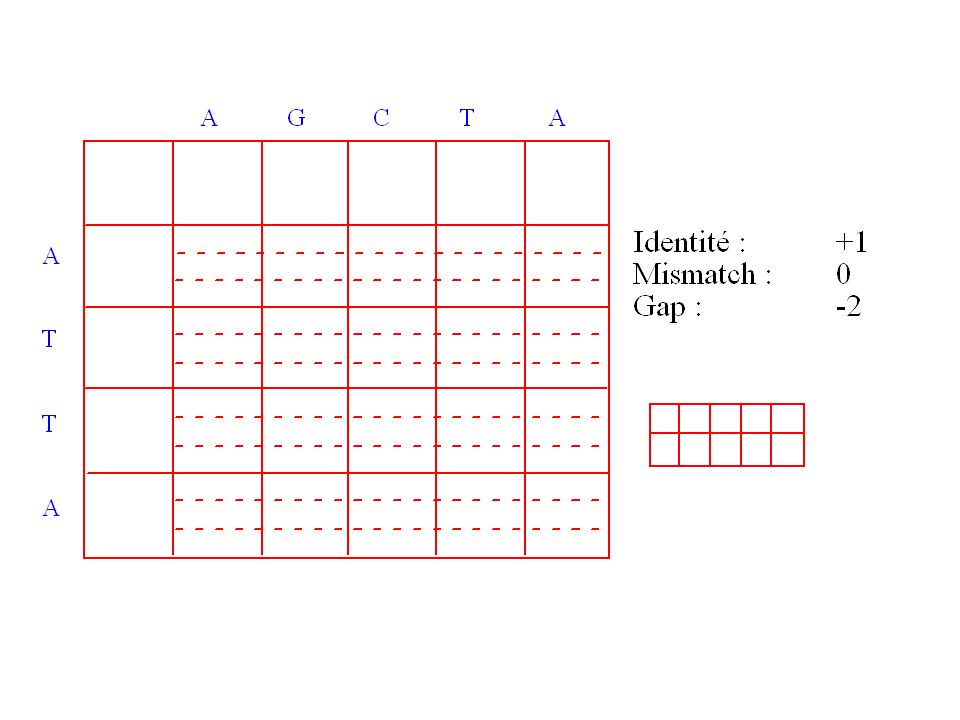
Définition de la matrice de chemins
Les alignements peuvent être représentés sous la forme d’une trajectoire dans une matrice de chemins. Pour chaque trajectoire on peut calculer un score et il faut donc trouver celle qui optimise ce score. Soit deux séquences A et B de longueurs respectives m et n définissant une matrice de chemin S. Dans chaque case de cette matrice on va stocker S(i, j), le score optimum de la trajectoire permettant d’arriver à cette case.
, le score optimum de la trajectoire permettant d’arriver à cette case.")
20
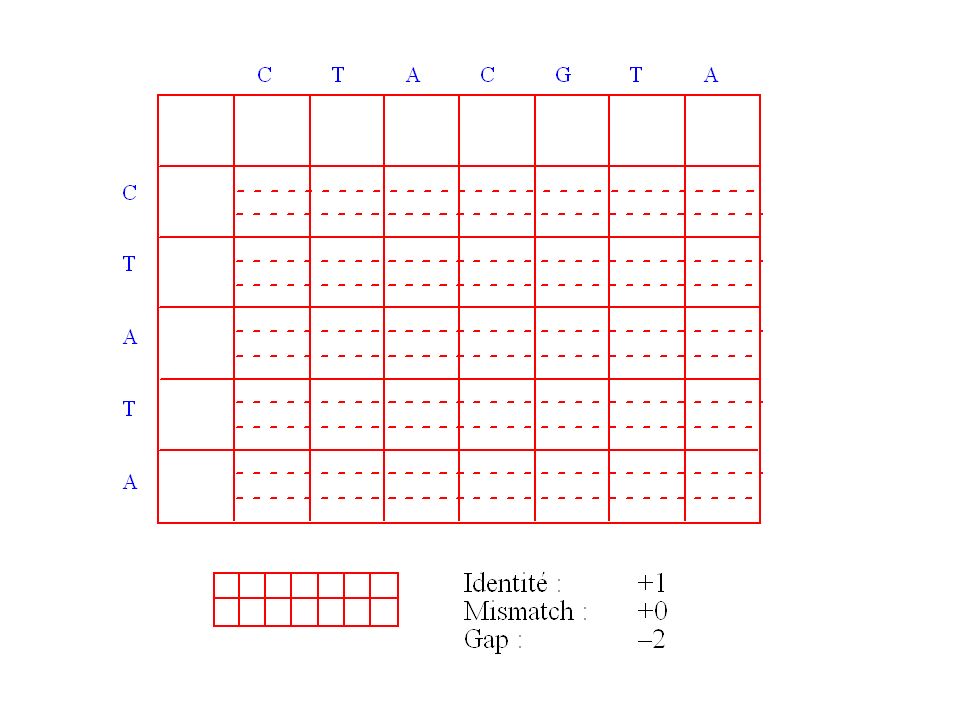
Exemple de matrice de chemin

21
Construction récursive de la matrice
Soit la case de coordonnées (i, j). Quelle que soit la trajectoire retenue, elle passera forcément par l’une des trois cases la précédant, de coordonnées (i–1, j), (i–1, j–1), (i, j–1). Supposons que l’on connaisse les scores optimums des trois cases précédentes, dans ce cas la valeur optimum du score dans la case (i, j) sera égale à : Needleman et Wunsh, 1970
. Quelle que soit la trajectoire retenue, elle passera forcément par l’une des trois cases la précédant, de coordonnées (i–1, j), (i–1, j–1), (i, j–1). Supposons que l’on connaisse les scores optimums des trois cases précédentes, dans ce cas la valeur optimum du score dans la case (i, j) sera égale à : Needleman et Wunsh,")
22
Bords de la matrice Les cases situées sur le bord du haut ou le bord gauche de la matrice ne possèdent plus le total requis de trois cases précédentes. Pour pallier ce problème on ajoute une ligne (0, j) et une colonne (i, 0) supplémentaires. Le balayage de la matrice ne se faisant plus qu’avec des indices ≥ 1 on ne rencontre plus de cases nécessitant un traitement particulier.
 et une colonne (i, 0) supplémentaires. Le balayage de la matrice ne se faisant plus qu’avec des indices ≥ 1 on ne rencontre plus de cases nécessitant un traitement particulier.")
23
Bords de la matrice (suite)
La ligne et la colonne supplémentaires doivent être initialisées pour pouvoir construire la matrice. Il existe plusieurs manières de faire selon la façon dont on veut comptabiliser les gains ou pertes d’éléments au niveau des extrémités. En particulier, il faut savoir si on veut pénaliser ou non les éléments terminaux non appariés (ce que l’on appelle les extrémités flottantes). - - - A T T C G T A T T C G T A T G A T T C G T A T G A T T C G T * * * * * * * * * * * *
 A T T C G T A T T C G T. A T G A T T C G T A T G A T T C G T. * * * * * * * * * * * *")
24
Bords de la matrice (fin)
Pénalisation des gaps terminaux Pas de pénalisation des gaps terminaux

26
Identité: +1 Mismatch: +0 Gap interne: -2 Gap terminal: +0

29
Alignement local (Smith-Waterman)
Initialisation des bords de la matrice de chemin à 0

30
Temps de calcul et occupation de la mémoire pour l'alignement de deux séquences de longueur n et m
Needleman-Wunsh Temps: O(n m) Espace mémoire: O(n m) Amélioration: éliminer les chemins qui s'éloignent trop de la diagonale Smith-Waterman Amélioration de Smith-Waterman Espace mémoire: O(n)
 Espace mémoire: O(n m) Amélioration: éliminer les chemins qui s éloignent trop de la diagonale. Smith-Waterman. Amélioration de Smith-Waterman. Espace mémoire: O(n)")
31
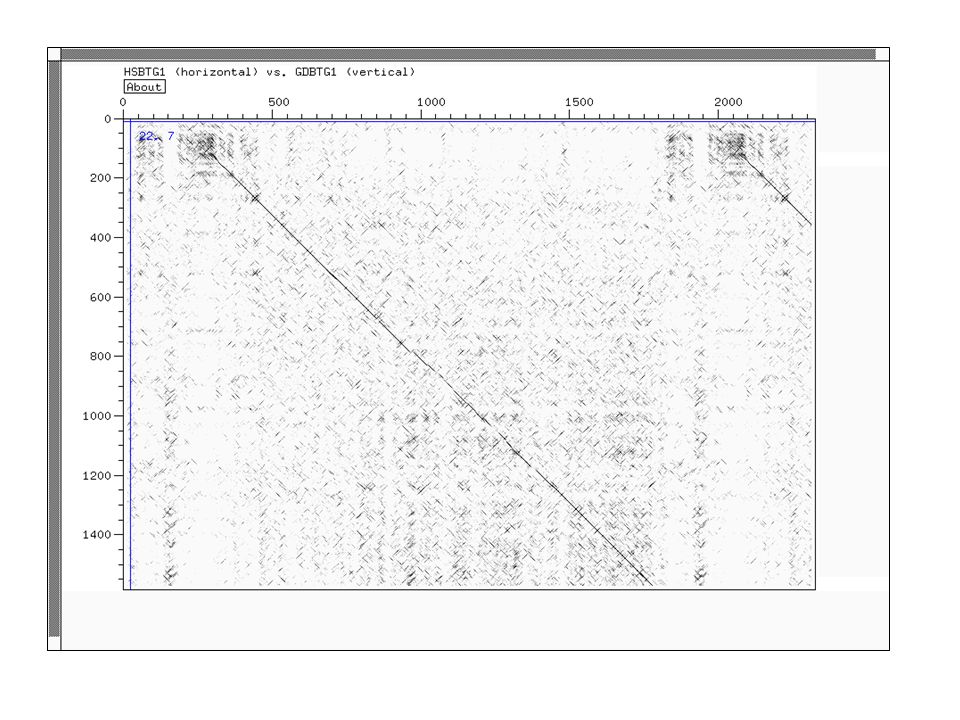
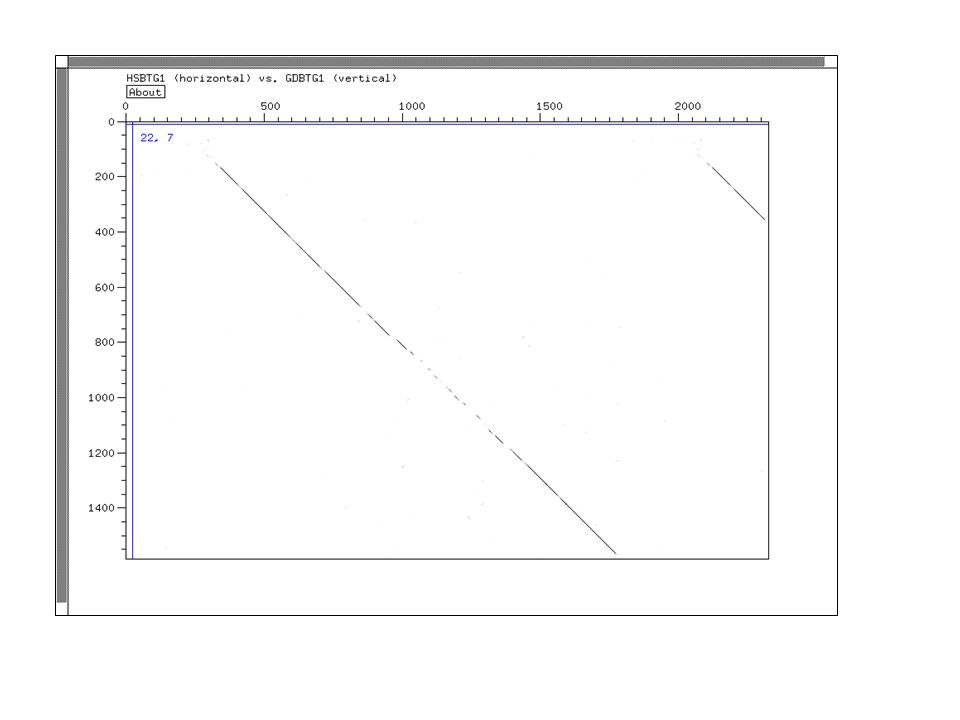
Dot Plot Représentation graphique de régions d'identité ou de similarité entre deux séquences Utilisation de fenêtres et de seuils pour réduire le bruit de fond Visualisation des inversion, duplications, palindromes

34
Recherche rapide de similarités dans les banques de séquences
Comparaison d'une séquence à toute une banque de données de séquences, comparaisons entre deux banques … Algorithmes exhaustifs (Smith-Waterman) DAP, BLITZ, SSEARCH, … Algorithmes basés sur des heuristiques FASTA 1 - recherche de ‘ k-tuplets ’ identiques 2 - alignement global, ancré sur la région similaire BLAST 1 - recherche de ‘ mots ’ similaires 2 - extension des blocs similaires
 DAP, BLITZ, SSEARCH, … Algorithmes basés sur des heuristiques. FASTA. 1 - recherche de ‘ k-tuplets ’ identiques. 2 - alignement global, ancré sur la région similaire. BLAST. 1 - recherche de ‘ mots ’ similaires. 2 - extension des blocs similaires.")
35
BLAST

36
Alignement par bloc ou alignement global : comparaison BLAST / FASTA

37
Stratégies de recherche de similarités: ADN ou protéine ?
Limites des recherches de similarité au niveau ADN Alphabet réduit (4 lettres) Dégénérescence du code génétique Mais … tout n'est pas codant régions régulatrices, ARN structuraux, ... Deux brins!
 Dégénérescence du code génétique. Mais … tout n est pas codant. régions régulatrices, ARN structuraux, ... Deux brins!")
38
Différentes versions de BLAST adaptées à différents problèmes
blastp: protéine/protéine blastn: ADN/ADN (utile pour non-codant) blastx: ADN-traduit/protéine (utile pour séquences codantes non-identifiées; plus sensible que blastn) tblastn: protéine/ADN-traduit (utile pour rechercher des homologues de gènes protéiques dans un génome non-entièrement annoté; plus sensible que blastn)
 blastx: ADN-traduit/protéine (utile pour séquences codantes non-identifiées; plus sensible que blastn) tblastn: protéine/ADN-traduit (utile pour rechercher des homologues de gènes protéiques dans un génome non-entièrement annoté; plus sensible que blastn)")
39
Choix de la matrice de substitutions
Différentes matrices de substitutions, adaptées à différentes distances évolutives BLOSUM 62: convient pour une large gamme de distances évolutives Combiner plusieurs matrices

40
Évaluation statistique de la similarité
Parmi les similarités qui ont été détectées, quelles sont celles qui reflètent des relations biologiquement importantes, quelles sont celles qui sont simplement dues au hasard ? Distribution des scores d'alignements locaux optimaux entre séquences non homologues Probabilité qu'une similarité de score S soit simplement due au hasard Nombre d'occurrences Score

41
Traitement du bruit de fond: filtres et masques
Similarités sans intérêt biologique Séquences de faible complexité (protéines, ADN): 40% des protéines ADN: microsatellites 15% du total des résidus exemple: CACACACACACACACACA Ala, Gly, Pro, Ser, Glu, Gln logiciels de filtrage: SEG, XNU, DUST RSPPR--KPQGPPQQEGNNPQGPPPPAGGNPQQPQAPPAGQPQGPP . ::: : :: : : ::::: : :: :.: :: : ::::: QGPPRPGNQQCPPPQGG--PQGPPRP--GNQQRP--PPQGGPQGPP Séquences abondantes 3000 Immunoglobulines dans GenBank 106 Alu, 105 L1 dans le génome humain logiciels de masquage: XBLAST, RepeatMasker
: 40% des protéines ADN: microsatellites. 15% du total des résidus exemple: CACACACACACACACACA. Ala, Gly, Pro, Ser, Glu, Gln. logiciels de filtrage: SEG, XNU, DUST. RSPPR--KPQGPPQQEGNNPQGPPPPAGGNPQQPQAPPAGQPQGPP. . ::: : :: : : ::::: : :: :.: :: : ::::: QGPPRPGNQQCPPPQGG--PQGPPRP--GNQQRP--PPQGGPQGPP. Séquences abondantes Immunoglobulines dans GenBank. 106 Alu, 105 L1 dans le génome humain. logiciels de masquage: XBLAST, RepeatMasker.")
42
Bilan: quelle approche adopter ?
algorithme matrices de substitution, pondération des gaps stratégie de recherche (nucléique, protéique) traitement du bruit de fond complétude des banques de données 1 - logiciel rapide, paramètres par défaut 2 - filtrage éventuel 3 - changement des paramètres (matrices, W, k, etc.) 4 - changement d'algorithme 5 - répéter la recherche régulièrement
 traitement du bruit de fond. complétude des banques de données. 1 - logiciel rapide, paramètres par défaut. 2 - filtrage éventuel. 3 - changement des paramètres (matrices, W, k, etc.) 4 - changement d algorithme. 5 - répéter la recherche régulièrement.")
43
Alignement multiple: programmation dynamique
La généralisation de l’algorithme précédent au traitement simultané de plus de deux séquences est théoriquement possible mais inexploitable en pratique. Pour un alignement de n séquences le nombre de chemins possibles pour chaque case est de 2n – 1. On a une croissance exponentielle du temps de calcul et de l'espace mémoire requis en fonction du nombre de séquences. Problème du choix d ’une fonction de score Utilisation de méthodes heuristiques.

44
Alignement progressif
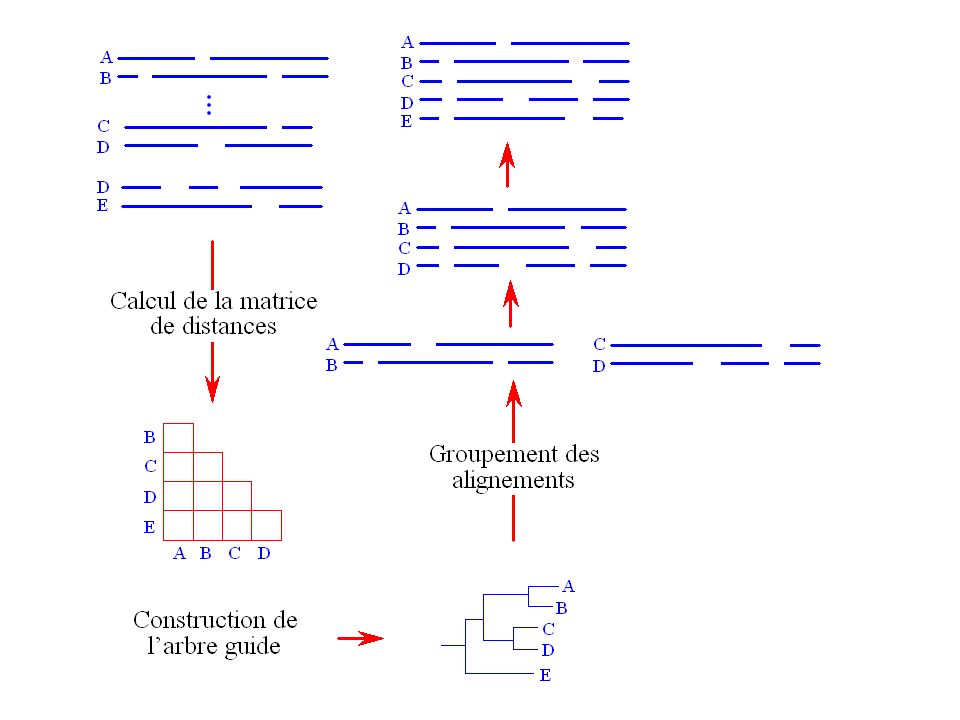
Approche consistant à construire itérativement l’alignement multiple en groupant des alignements de paires de séquences. Ce genre de méthodes comporte trois étapes : L’alignement des paires de séquences. Le groupement des séquences. Le groupement des alignements (alignement progressif). CLUSTAL (Thompson et al., 1994), le programme d’alignements multiples le plus utilisé à l’heure actuelle utilise cette approche.
. CLUSTAL (Thompson et al., 1994), le programme d’alignements multiples le plus utilisé à l’heure actuelle utilise cette approche.")
46
Pénalités initiales pour les gaps
CLUSTAL utilise une fonction de pénalité linéaire pour les gaps. De plus, les valeurs initiales de do et de sont corrigées en fonction de nombreux facteurs : Le degré de similarité entre les séquences : do µ %identité(A, B) La longueur des séquences : do µ log[min(m, n)] La différence de longueur entre les deux séquences : de µ |log[n/m]| Ces pondérations sont prises en compte au moment de l’alignement des paires de séquences.
 La longueur des séquences : do µ log[min(m, n)] La différence de longueur entre les deux séquences : de µ |log[n/m]| Ces pondérations sont prises en compte au moment de l’alignement des paires de séquences.")
47
Pénalités en fonction de la position
CLUSTAL introduit également des pondérations qui sont dépendantes de la position des gaps. Diminution de la pénalité à l’emplacement de gaps préexistants. Augmentation de la pénalité au voisinage (8 résidus) de gaps préexistants. Réduction de la pénalité au niveau de régions contenant des suites d’acides aminés hydrophiles (≥ 5 résidus). Modification spécifiques en fonction des acides aminés présents (e.g., la pénalité est plus faible avec Gly, Asn, Pro). Ces pondérations sont prises en compte au moment du groupement des alignements.
 de gaps préexistants. Réduction de la pénalité au niveau de régions contenant des suites d’acides aminés hydrophiles (≥ 5 résidus). Modification spécifiques en fonction des acides aminés présents (e.g., la pénalité est plus faible avec Gly, Asn, Pro). Ces pondérations sont prises en compte au moment du groupement des alignements.")
48
Alignement progressif: pas toujours optimal
Un seul des ces trois alignements est optimal

49
Global Alignments, Block alignments

50
Dialign Morgenstern et al. 1996 PNAS 93:12098
Search for similar blocks without gap Select the best combination of consistent similar blocks (uniforms or not) : heuristic (Abdeddaim 1997) Alignment anchored on blocks Slower than progressive alignment, but better when sequences contain large indels Do not try to align non-conserved regions
 : heuristic (Abdeddaim 1997) Alignment anchored on blocks. Slower than progressive alignment, but better when sequences contain large indels. Do not try to align non-conserved regions.")
51
Local Multiple Alignments
MEME MATCH-BOX PIMA

52
Overview ClustalW Dialign T-coffee MEME

53
Éditeur d ’alignement multiple

54
Special cases Alignment of coding DNA sequences
L F L F CTT TTC CTT TTC CTC CTC L L alignment of protein sequences back-translation of the protein alignment into a DNA alignment Alignment cDNA / genomic DNA: SIM4 Alignment protein / genomic DNA : GeneWise

55
Limits of pairwise comparison (BLAST, FASTA, ...)
Seq A CGRRLILFMLATCGECDTDSSE … HICCIKQCDVQDIIRVCC :: : ::: :: : : Insulin CGSHLVEALYLVCGERGFFYTP … EQCCTSICSLYQLENYCN ::: : : : :: : : Seq B YQSHLLIVLLAITLECFFSDRK … KRQWISIFDLQTLRPMTA Pairwise comparison: Insulin / Seq A : 25% identity Insulin / Seq B : 25% identity

56
Insulin gene family: sequence alignment
B-chain A-chain INSL4 Q ELRGCGPRFGKHLLSYCPMPEKTFTTTPGG...[x] SGRHRFDPFCCEVICDDGTSVKLCT INSL3 P REKLCGHHFVRALVRVCGGPRWSTEA [x] AAATNPARYCCLSGCTQQDLLTLCPY RLN1 P VIKLCGRELVRAQIAICGMSTWS [x] PYVALFEKCCLIGCTKRSLAKYC BBXA P VHTYCGRHLARTLADLCWEAGVD [x] GIVDECCLRPCSVDVLLSYC BBXB P ARTYCGRHLADTLADLCF--GVE [x] GVVDECCFRPCTLDVLLSYCG BBXC P SQFYCGDFLARTMSILCWPDMP [x] GIVDECCYRPCTTDVLKLYCDKQI BBXD P GHIYCGRYLAYKMADLCWRAGFE [x] GIADECCLQPCTNDVLLSYC LIRP P VARYCGEKLSNALKLVCRGNYNTMF [x] GVFDECCRKSCSISELQTYCGRR MIP I P RRGVCGSALADLVDFACSSSNQPAMV [x] QGTTNIVCECCMKPCTLSELRQYCP MIP II P PRGICGSNLAGFRAFICSNQNSPSMV [x] QRTTNLVCECCFNYCTPDVVRKYCY MIP III P PRGLCGSTLANMVQWLCSTYTTSSKV [x] ESRPSIVCECCFNQCTVQELLAYC MIP V P PRGICGSDLADLRAFICSRRNQPAMV [x] QRTTNLVCECCYNVCTVDVFYEYCY MIP VII P PRGLCGNRLARAHANLCFLLRNTYPDIFPR...[x]86 ..EVMAEPSLVCDCCYNECSVRKLATYC ILP P AEYLCGSTLADVLSFVCGNRGYNSQP [x] GLVEECCYNVCDYSQLESYCNPYS INS P NQHLCGSHLVEALYLVCGERGFFYTPKT.....[x] GIVEQCCTSICSLYQLENYCN IGF1 P PETLCGAELVDALQFVCGDRGFYF [x] GIVDECCFRSCDLRRLEMYCAPLK IGF2 P SETLCGGELVDTLQFVCGDRGFYF [x] GIVEECCFRSCDLALLETYCATPA * * ** * *

57
Biomolecular Sequence Motif Descriptors
Exact word: e.g. EcoRI restriction site GAATTC Consensus: e.g. TATA box: TATAWAWR Regular expression: e.g. insulins PROSITE pattern C-C-{P}-x(2-4)-C-[STDNEKPI]-x(3)-[LIVMFS]-x(3)-C Weight matrix: position-specific weighting of substitutions Generalised profiles (hidden markov models) : position-specific weighting of substitutions and indels
-C-[STDNEKPI]-x(3)-[LIVMFS]-x(3)-C. Weight matrix: position-specific weighting of substitutions. Generalised profiles (hidden markov models) : position-specific weighting of substitutions and indels.")
58
Example of weight matrix
Splice donnor sites of vertebrates: frequency (%) of the four bases at each position log transformation weight matrix Base Position A C G T Cons. M A G G T R A G T
 of the four bases at each position. log transformation weight matrix. Base Position A C G T Cons. M A G G T R A G T.")
59
Searching for distantly related homologues in sequence databases
1- search for homologues (e.g. BLAST) 2- align homologues (e.g. CLUSTAL, MEME) 3- compute a profile from the multiple alignment 4- compare the profile to a sequence database (e.g. MAST, pfsearch) pfsearch: MEME/MAST:
 2- align homologues (e.g. CLUSTAL, MEME) 3- compute a profile from the multiple alignment. 4- compare the profile to a sequence database (e.g. MAST, pfsearch) pfsearch: MEME/MAST:")
60
PSI-BLAST Position-Specific Iterated BLAST 1- classical BLAST search
2- compute a profile with significant BLAST hits 3- BLAST search based on the profile 4 -repeat steps 2-3 up to convergence More sensitive than Smith-Waterman 40 times faster

61
Comparison of a sequence to a database of protein motifs
Databases: PROSITE, PFAM, PRODOM, …, INTERPRO Search tools: ProfileScan :

Présentations similaires