Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Syndrome de Guillain-Barré RENALDO Florence SENP 2013
Guillain G, Barré JA, Ströhl A. Sur un syndrome de radiculonévrite avec hyperalbuminose du liquide céphalo-rachidien sans réaction cellulaire: remarques sur les caractères cliniques et graphiques des réflexes tendineux. Bulletins et mémoires de la Société des Médecins des Hôpitaux de Paris 1916; 40: Syndrome de Guillain-Barré RENALDO Florence SENP 2013

2
Un peu d’histoire… Guillain, Barré et Strohl, 1916:clinique + dissoc alb cyto; 2 soldats, paralysie ascen aréflexie, guérison spont; dgn diff de polyomyélite Fisher M. An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). N Engl J Med 1956; 255:57-65 Asbury et al., 1969: polyneuropathie démyélinisante inflammatoire aigue (électrophysio, neuropath) Feasby et al. en 1986: 1ere forme axonale décrite, puis largement étudiée en Chine Winer JB et al. 1988, Jacobs BC et al. 1998: signification de l’infection précedant GB. infection à C.jéjuni, CMV, EBV, Mycoplasma significativement + fréquente chez 154 patients GB que chez les 154 controles; assoc C.jejuni et anti-GM1, anti-GD1b/ CMV anti-GM2 Rees et al. 1995: prospectif, 96 patients GB + 7 MF sur 2ans: association épidémiologique significative entre infection récente à C.jejuni et sd de GB ou MF; dégénerescence axonale, récupération lente, séquelles sévères.
. N Engl J Med 1956; 255: Asbury et al., 1969: polyneuropathie démyélinisante inflammatoire aigue (électrophysio, neuropath) Feasby et al. en 1986: 1ere forme axonale décrite, puis largement étudiée en Chine. Winer JB et al. 1988, Jacobs BC et al. 1998: signification de l’infection précedant GB. infection à C.jéjuni, CMV, EBV, Mycoplasma significativement + fréquente chez 154 patients GB que chez les 154 controles; assoc C.jejuni et anti-GM1, anti-GD1b/ CMV anti-GM2. Rees et al. 1995: prospectif, 96 patients GB + 7 MF sur 2ans: association épidémiologique significative entre infection récente à C.jejuni et sd de GB ou MF; dégénerescence axonale, récupération lente, séquelles sévères.")
3
Un peu d’histoire… Guillain G, Barré JA, Ströhl A. Sur un syndrome de radiculonévrite avec hyperalbuminose du liquide céphalo-rachidien sans réaction cellulaire: remarques sur les caractères cliniques et graphiques des réflexes tendineux. Bulletins et mémoires de la Société des Médecins des Hôpitaux de Paris 1916; 40: Fisher M. Syndrome of ophthalmoplegia, ataxia and areflexia. N Engl J Med 1956; 255: 57– 65. Asbury AK, Arnason BG, Adams RD. The inflammatory lesion in idiopathic polyneuritis. Its role in pathogenesis. Medicine 1969; 48: 173–215. Feasby et al. en 1986: 1ere forme axonale décrite, puis largement étudiée en Chine Winer JB, Hughes RAC, Anderson MJ, Jones DM, Kangro H, Watkins RP. A prospective study of acute idiopathic neuropathy. II: antecedent events. J Neurol Neurosurg Psychiatry 1988; 51:613-8. Jacobs BC, Rothbarth PH, van der Meché FGA, Et al. The spectrum of antecedent infections in Guillain-Barré syndrome: a case-control study. Neurology 1998; 51:1110-5 Rees JH, Soudain SE, Gregson NA, Hughes RAC. Campylobacter jejuni infection and Guillain-Barré syndrome. N Engl J Med 1995; 333:1374-9 Guillain, Barré et Strohl, 1916:clinique + dissoc alb cyto; 2 soldats, paralysie ascen aréflexie, guérison spont; dgn diff de polyomyélite Fisher M. Asbury et al., 1969: polyneuropathie démyélinisante inflammatoire aigue (électrophysio, neuropath) Feasby et al. en 1986: 1ere forme axonale décrite, puis largement étudiée en Chine Winer JB et al. 1988, Jacobs BC et al. 1998: signification de l’infection précedant GB. infection à C.jéjuni, CMV, EBV, Mycoplasma significativement + fréquente chez 154 patients GB que chez les 154 controles; assoc C.jejuni et anti-GM1, anti-GD1b/ CMV anti-GM2 Rees et al. 1995: prospectif, 96 patients GB + 7 MF sur 2ans: association épidémiologique significative entre infection récente à C.jejuni et sd de GB ou MF; dégénerescence axonale, récupération lente, séquelles sévères. 3
 Feasby et al. en 1986: 1ere forme axonale décrite, puis largement étudiée en Chine. Winer JB et al. 1988, Jacobs BC et al. 1998: signification de l’infection précedant GB. infection à C.jéjuni, CMV, EBV, Mycoplasma significativement + fréquente chez 154 patients GB que chez les 154 controles; assoc C.jejuni et anti-GM1, anti-GD1b/ CMV anti-GM2. Rees et al. 1995: prospectif, 96 patients GB + 7 MF sur 2ans: association épidémiologique significative entre infection récente à C.jejuni et sd de GB ou MF; dégénerescence axonale, récupération lente, séquelles sévères. 3.")
4
Sd de Guillain-Barré Polyradiculonévrite aigue, acquise
Définition: atteinte inflammatoire post-infectieuse, auto-immune, du SNP, aigue et acquise Epidémiologie: 0.5 à 1/ cas par an pour <18ans. âge moyen 6,3ans (11m-17,7a) ; inhabituel chez <2ans M:1,5F Physiopathologie: Réponse immune aberrante post-infectieuse, responsable de lésions du SNP
 ; inhabituel chez <2ans. M:1,5F. Physiopathologie: Réponse immune aberrante post-infectieuse, responsable de lésions du SNP.")
5
P.Anas 1 Filllette de 6ans hosp pour troubles de la marche aigus
Pas d’atcd familiaux , 1ere enfant Nce à terme eutrophe, ictère néonatal traité par photothérapie Retard de langage léger, orthophonie GSM

6
P.Anas 2 Angine suivie (1mois après) par douleurs des MI. Cs d’orthopédie, ex neurologique normal Cs le lendemain pour persistance des douleurs, prurit. BS : CPK à 308UI/L, isolées=> myosite virale? Paresthésies des MI progressives, perte de la marche à J3. Aux urgences: areflexie des 4 mb ponction lombaire en faveur d’une dissociation albumino- cytologique (5cell, prot 1,24g/l) EMG polyradiculonévrite aigue type démyélinisante (aug des latences distales, dim des v de conduction sensitives et motrices, amplitudes motrices éffondrées, sensitives conservées) Trt par tégélines : 1g/kg/j sur 2jours
 EMG polyradiculonévrite aigue type démyélinisante (aug des latences distales, dim des v de conduction sensitives et motrices, amplitudes motrices éffondrées, sensitives conservées) Trt par tégélines : 1g/kg/j sur 2jours.")
7
P.Anas 3 Transfert en réanimation à J10 devant: évolution rapidement progresssive de tétraplégie flasque, atteinte des paires craniennes (PF, absence de réflexe nauséeux, stase salivaire), atteinte respiratoire (hypoventilation), troubles dysautonomiques (HTA, épisodes de tachycardie supra- ventriculaire/bradycardie ) Intubation-ventilation assistée de J10 à J15, nutrition entérale
, atteinte respiratoire (hypoventilation), troubles dysautonomiques (HTA, épisodes de tachycardie supra- ventriculaire/bradycardie ) Intubation-ventilation assistée de J10 à J15, nutrition entérale.")
8
P.Anas 4 Persistance d’une HTA (loxen IV puis Adalate)
Plasmaphérèses X4 2eme cure de tégélines Amélioration progressive: ventilation spont en AA à J22 Reprise alimentation per os à J21

9
P.Anas 5 PCR CMV + (sang) PCR mycoplasme, sérologie Lyme, syphilis, campylobacter, hépatites A, B, C : - Anti gangliosides (anti GM2)
")
10
P.Anas 6 Évolution 2 mois après la fin du trt:
Marche avec aide possible Hyperesthésie et diplopie quasi résolutives ROT encore abolis, pas de R nauséeux Diminution des anti-GM2 Évolution à 8mois: - difficultés motrices persistantes

11
Sd douloureux (90%): myalgies+++, radiculalgies, paresthésies…
Sd de Guillain-Barré La clinique 1j-Xm 7j-28m 1-30j Forme classique: paralysie flasque rapidement progressive (<4sem), acendante sym avec aréflexie Sd douloureux (90%): myalgies+++, radiculalgies, paresthésies… Déficit de la force musculaire : Refus de la marche, dandinement, instabilité (ataxie)… Hypotonie =>parésie/paralysie flasque proximale puis extrémités ROT abolis Pas de sd pyramidal, d’amyotrophie, troubles sensitifs objectifs très rares Atteinte respiratoire (47%), Atteinte des paires craniennes (2/3) Dysautonomie (2/3): dysrégulation FC et ou PA, flush, sudation, troubles digestifs, vésico- sphinctériens, anomalie pupillairett Infection (respiratoire, ORL, digestives) <1mois (2/3)
, acendante sym avec aréflexie. Sd douloureux (90%): myalgies+++, radiculalgies, paresthésies… Déficit de la force musculaire : Refus de la marche, dandinement, instabilité (ataxie)… Hypotonie =>parésie/paralysie flasque proximale puis extrémités. ROT abolis. Pas de sd pyramidal, d’amyotrophie, troubles sensitifs objectifs très rares. Atteinte respiratoire (47%), Atteinte des paires craniennes (2/3) Dysautonomie (2/3): dysrégulation FC et ou PA, flush, sudation, troubles digestifs, vésico- sphinctériens, anomalie pupillairett. Infection (respiratoire, ORL, digestives) <1mois (2/3)")
12
Principales bacteries: campylobacter jejuni, mycoplasme,
Sd de Guillain-Barré Les facteurs déclenchants Principales bacteries: campylobacter jejuni, mycoplasme, Prinvipaux virus: CMV, VZV, EBV, HIV +/- Vaccins (antigrippal) Les lésions démyélinisantes du SGB seraient en rapport avec la production et le passage dans les espaces endoneuraux d'anticorps dirigés contre certains antigènes de la myéline, dont la nature n'a pas encore été établie chez l'homme. L’infection virale déclenche une immunisation croisée contre les antigènes du système nerveux périphérique. Cette participation humorale est confirmée par la démyélinisation observée "in vivo" chez l'animal après injection de sérum de patients atteints.
 Les lésions démyélinisantes du SGB seraient en rapport avec la production et le passage dans. les espaces endoneuraux d anticorps dirigés contre certains antigènes de la myéline, dont la. nature n a pas encore été établie chez l homme. L’infection virale déclenche une immunisation. croisée contre les antigènes du système nerveux périphérique. Cette participation humorale est. confirmée par la démyélinisation observée in vivo chez l animal après injection de sérum de. patients atteints.")
13
PL : dissociation albuminocytologique
Sd de Guillain-Barré Diagnostic Clinique PL : dissociation albuminocytologique hyperprotéinorrachie (J3-J10) <6g/l cellularité nle (< 50 élémts/mm3) EMG : Neuropathies démyélinisantes : ralentissement diffus des vitesses de conduction motrices et sensitives, allongement des ondes tardives F et des latences distales motrices. Possible aspect de bloc de conduction Neuropathies axonales : les vitesses sont normales ou modérément ralenties, la baisse d'amplitude de la réponse motrice et du potentiel sensitif renseigne sur le nombre d’axones fonctionnels. - Neuropathies démyélinisantes : ralentissement diffus des vitesses de conduction motrices et sensitives, allongement des ondes tardves F et des latences distales motrices. Il peut exister un aspect de bloc de conduction moteur (rapport diminué entre l’amplitude obtenue par stimulation proximale et l’amplitude obtenue par stimulation distale) notamment dans les polyradiculonévrites. Ces anomalies sont localisées dans les mononeuropathies multiples et les compressions. - Neuropathies axonales : les vitesses sont normales ou modérément ralenties, la baisse d'amplitude de la réponse motrice et du potentiel sensitif renseigne sur le nombre d’axones fonctionnels. La mesure des vitesses de conductions objective : - une réduction des rapports des réponses motrices obtenues par stimulation proximale et distale (bloc de conduction). - une augmentation de la latence distale motrice (atteinte des fibres les plus rapides). - un ralentissement des vitesses de conduction. - l’observation précoce d’une démyélinisation proximale par allongement des latences des ondes tardives F. Il n’y a pas de parallélisme entre le degré de la paralysie et le ralentissement des VCM. La multiplicité des lésions impose l’examen sur plusieurs troncs nerveux. Enfin, les potentiels d'action sensitifs sont souvent normaux au début. L’EMG de détection détecte les éléments d’une atteinte axonale associée de mauvais pronostic fonctionnel : fibrillations et potentiels de dénervation au repos, sommation temporelle à l’effort (Potentiels déchargeant à une fréquence anormale).
 <6g/l. cellularité nle (< 50 élémts/mm3) EMG : Neuropathies démyélinisantes : ralentissement diffus des vitesses de conduction motrices et sensitives, allongement des ondes tardives F et des latences distales motrices. Possible aspect de bloc de conduction. Neuropathies axonales : les vitesses sont normales ou modérément ralenties, la baisse d amplitude de la réponse motrice et du potentiel sensitif renseigne sur le nombre d’axones fonctionnels. - Neuropathies démyélinisantes : ralentissement diffus des vitesses de conduction. motrices et sensitives, allongement des ondes tardves F et des latences distales motrices. Il. peut exister un aspect de bloc de conduction moteur (rapport diminué entre l’amplitude. obtenue par stimulation proximale et l’amplitude obtenue par stimulation distale) notamment dans les polyradiculonévrites. Ces anomalies sont localisées dans les mononeuropathies multiples et les compressions. - Neuropathies axonales : les vitesses sont normales ou modérément ralenties, la baisse. d amplitude de la réponse motrice et du potentiel sensitif renseigne sur le nombre d’axones. fonctionnels. La mesure des vitesses de conductions. objective : - une réduction des rapports des réponses motrices obtenues par stimulation proximale et. distale (bloc de conduction). - une augmentation de la latence distale motrice (atteinte des fibres les plus rapides). - un ralentissement des vitesses de conduction. - l’observation précoce d’une démyélinisation proximale par allongement des latences des. ondes tardives F. Il n’y a pas de parallélisme entre le degré de la paralysie et le ralentissement des VCM. La. multiplicité des lésions impose l’examen sur plusieurs troncs nerveux. Enfin, les potentiels. d action sensitifs sont souvent normaux au début. L’EMG de détection détecte les éléments d’une atteinte axonale associée de mauvais. pronostic fonctionnel : fibrillations et potentiels de dénervation au repos, sommation. temporelle à l’effort (Potentiels déchargeant à une fréquence anormale).")
14
FO : rarement oedème papillaire
Sd de Guillain-Barré Diagnostic FO : rarement oedème papillaire IRM moëlle: prise de contraste des racines, du cône terminal - Neuropathies démyélinisantes : ralentissement diffus des vitesses de conduction motrices et sensitives, allongement des ondes tardves F et des latences distales motrices. Il peut exister un aspect de bloc de conduction moteur (rapport diminué entre l’amplitude obtenue par stimulation proximale et l’amplitude obtenue par stimulation distale) notamment dans les polyradiculonévrites. Ces anomalies sont localisées dans les mononeuropathies multiples et les compressions. - Neuropathies axonales : les vitesses sont normales ou modérément ralenties, la baisse d'amplitude de la réponse motrice et du potentiel sensitif renseigne sur le nombre d’axones fonctionnels. La mesure des vitesses de conductions objective : - une réduction des rapports des réponses motrices obtenues par stimulation proximale et distale (bloc de conduction). - une augmentation de la latence distale motrice (atteinte des fibres les plus rapides). - un ralentissement des vitesses de conduction. - l’observation précoce d’une démyélinisation proximale par allongement des latences des ondes tardives F. Il n’y a pas de parallélisme entre le degré de la paralysie et le ralentissement des VCM. La multiplicité des lésions impose l’examen sur plusieurs troncs nerveux. Enfin, les potentiels d'action sensitifs sont souvent normaux au début. L’EMG de détection détecte les éléments d’une atteinte axonale associée de mauvais pronostic fonctionnel : fibrillations et potentiels de dénervation au repos, sommation temporelle à l’effort (Potentiels déchargeant à une fréquence anormale).
 notamment dans les polyradiculonévrites. Ces anomalies sont localisées dans les mononeuropathies multiples et les compressions. - Neuropathies axonales : les vitesses sont normales ou modérément ralenties, la baisse. d amplitude de la réponse motrice et du potentiel sensitif renseigne sur le nombre d’axones. fonctionnels. La mesure des vitesses de conductions. objective : - une réduction des rapports des réponses motrices obtenues par stimulation proximale et. distale (bloc de conduction). - une augmentation de la latence distale motrice (atteinte des fibres les plus rapides). - un ralentissement des vitesses de conduction. - l’observation précoce d’une démyélinisation proximale par allongement des latences des. ondes tardives F. Il n’y a pas de parallélisme entre le degré de la paralysie et le ralentissement des VCM. La. multiplicité des lésions impose l’examen sur plusieurs troncs nerveux. Enfin, les potentiels. d action sensitifs sont souvent normaux au début. L’EMG de détection détecte les éléments d’une atteinte axonale associée de mauvais. pronostic fonctionnel : fibrillations et potentiels de dénervation au repos, sommation. temporelle à l’effort (Potentiels déchargeant à une fréquence anormale).")
15
Spectre phénotypique : AIDP AMAN, AMSAN Sd de Miller-Fisher (1956)
Sd de Guillain-Barré Variants cliniques Spectre phénotypique : AIDP AMAN, AMSAN Sd de Miller-Fisher (1956) Bickerstaff brainstem encephalitis (1951) pharyngo-cervical-brachial variant polynévrite des N craniens -De + en + clair que le BG ne constitue pas une seule entité mais représente plutôt un groupe hétérogène sur le plan clinique et neuropathologique, d’atteinte aigue du SNP voire du SNC -MF: ac contre épitopes des ganglions des racines sensitives, neurones du cervelet, N oculomoteur -il est important de reconnaître les cas atypiques afin d’anticiper les complications et d’adapter le trt
 Bickerstaff brainstem encephalitis (1951) pharyngo-cervical-brachial variant. polynévrite des N craniens. -De + en + clair que le BG ne constitue pas une seule entité mais représente plutôt un groupe hétérogène sur le plan clinique et neuropathologique, d’atteinte aigue du SNP voire du SNC. -MF: ac contre épitopes des ganglions des racines sensitives, neurones du cervelet, N oculomoteur. -il est important de reconnaître les cas atypiques afin d’anticiper les complications et d’adapter le trt.")
16
85% des GB en Europe, USA, Australie
AIDP AMAN AMSAN MFS BBE fréquence 85% des GB en Europe, USA, Australie 30-47% des GB en Chine, japon, Am du sud symptômes Paralysie flasque prog, areflexie, PC + Pas de def sensitif Tetraparesie et déficit sensitif sévères PC Ophtalmoplégie Ataxie Aréflexie Diplopie, anisocorie, ptosis MFS + Troubles de conscience +sd pyramidal dysautonomie +++++ EMG-VCN VCN, onde F, bloc de conduction, LDM PAM PAM, PAS PAS N ou dim LCR Diss alb-cyt + + ou N infection Auto-AC CMV GM2 Camp.jejuni GM1, GD1a, GM1b, GalNAc –GD1a GQ1b, GD3, GT1a GQ1b évolution Récupération lente, forme + sévère Favorable moins sévère

17
Atteinte respiratoire initiale dont sévèrité > déficit moteur
Sd de Guillain-Barré Douter… Atteinte respiratoire initiale dont sévèrité > déficit moteur Signes sensitifs initiaux dont sévérité > signes moteurs Troubles vésico-sphinctériens d’emblée Fièvre inaugurale Niveau sensitif Progression lente, peu sévère Asymétrie persistante Hypercellularité: >50.106/l, cellules anormales

18
L. Hugo

19
L. Hugo Nce 40SA , circulaire cordon
2280g, 51.5cm, 34.5cm, (M) , Apgar 10/10 Eczema modéré, otite à 5m Dev PM N RCH chez le papa, asthme et allergie GP paternel 1er enfant, parents non apparenté
 , Apgar 10/10. Eczema modéré, otite à 5m. Dev PM N. RCH chez le papa, asthme et allergie GP paternel. 1er enfant, parents non apparenté.")
20
L. Hugo : HDM Début juin (8mois): cs multiples aux urgences pour constipation opiniâtre et refus + toux aux liquides À j7 d’évolution: hypotonie globale avec perte de la station assise, puis malaise brutal post prandial avec pâleur gal, rougeur conjonctivale, => hospitalisation Examen: bon contact mais tétraparésie flasque, aréflexie, raideur rachidienne marquée, douloureux, refuse la position assise

21
L. Hugo : explorations 1 Biologie standard, infectieux, métabolique: N
IRM cérébrale: kyste de la poche de Blake non patho => Transfert Hôpital Trousseau

22
L. Hugo : dans le service Bon contact
Encombrement VAS avec respiration abdominale, faible ampliation thoracique, ausc N Pas de trouble OM, ex des PC N Tétraplégie flasque, aréflexie RCA+ Rachis très douloureux et raide à la mobilisation Abdo: ras cutané: ras

23
Quels examens??? Devant tableau rapidement progressif:
Tétraplégie flasque aréflexique, troubles de la déglutition, atteinte respiratoire modérée, troubles sphinctériens, raideur rachis ????

24
L. Hugo : explorations 2 TDM puis IRM médullaire:
Pas de tumeur (os, moëlle) Pas de myélite Prise de contraste des racines nerveuses, ++cone terminal = polyradiculonévrite aigue PL: dissociation albumino-cytologique (2 elmts, prot 2.33g/l, gluc 3mmol/l R=0.8, culture stérile, pas de Σ intrathécale) EMG: neuropathie aigue SM démyélinisante sans atteinte axonale (diminution des V de conductions)
 Pas de myélite. Prise de contraste des racines nerveuses, ++cone terminal. = polyradiculonévrite aigue. PL: dissociation albumino-cytologique. (2 elmts, prot 2.33g/l, gluc 3mmol/l R=0.8, culture stérile, pas de Σ intrathécale) EMG: neuropathie aigue SM démyélinisante sans atteinte axonale (diminution des V de conductions)")
25
L. Hugo : évolution 1 À J15: aggravation rapide, extension du déficit moteur, fausses routes salivaires, pauses respiratoires brèves, majoration du syndrome douloureux => transfert en USI- réanimation pour surveillance rapprochée Tégélines IV 1g/kg 3j

26
L. Hugo : évolution 2 Neutropénie 560->260/mm3
Anémie microcytaire 9.5g/dl, VGM 63fl Carence martiale => Myélogramme Phénotypage lymphocytaire, sérologie VIH Bilan AI= N

27
L. Hugo : évolution 3 Régression des troubles après trt, douleur sous antalgiques (Nubain, Doliprane) J30: reprise de l ’alimentation, tenue de tête difficile, pas de tenue assise, saisit les objets et les passe d’une main à l’autre, lève les bras, pousse sur ses MI NFS normale le 05/07 soit j35

28
L. Hugo : bilan étiologique
Sérologie campylobacter Jejuni - Coproculture – Sérologie EBV, CMV, mycoplasma pneumoniae : - Avis hémato: neutropénie virale

29
L. Hugo : 20mois Examen clinique N Développement PM N
EMG-VCN: N (v motrices et sensitives)
")
30
Au total: L. Hugo Tableau sévère de polyradiculonévrite aigue de type démyélinisant (AIDP) avec déficit moteur complet, troubles de la déglutition, atteinte respiratoire, raideur rachidienne, douleurs Contexte infectieux non étiqueté Évolution favorable sous trt, récupération complète après 12m
 avec déficit moteur complet, troubles de la déglutition, atteinte respiratoire, raideur rachidienne, douleurs. Contexte infectieux non étiqueté. Évolution favorable sous trt, récupération complète après 12m.")
31
J.-J. Lin et al. / Pediatric Neurology 47 (2012) 91-96
 91-96")
32
C.Isa Parents d’origine chinoise Pas d’antécédents
Patiente de 12ans. Voyage en Chine: épisode de diarrhée À M1: troubles de déglutition, voix nasonnée, chutes Examen: déficit de la force musculaire prédominant en proximal (4 mb) ; aréflexie aux MI; hypotonie Biologie N; PL N; scanner cérébral N; absence de toxiques; recherches infectieuses sg et LCR -
 ; aréflexie aux MI; hypotonie. Biologie N; PL N; scanner cérébral N; absence de toxiques; recherches infectieuses sg et LCR -")
33
C.Isa Suspicion de sd de GB =>cure de tégélines

34
C.Isa 2e PL: 9 éléments EMG-VCN: tableau d’atteinte pre-synaptique; pas de bloc de conduction; pas d’atteinte axonale. Recherche de toxine botulique dans le sang et dans le selles: - Bilan auto-immun : - IRM cérébrale et médullaire: N 2e EMG-VCN: V normales; latences normales; certaines ondes F non recueillies. Recueil d’un incrément sur certains couples moteurs évocateur de bloc neuromusculaire pré- synaptique

35
C.Isa Quel DGN?????? Culture de clostridium botulinum dans les selles +++ Quasi disparition des symptomes à M3

36
Diagnostic différentiel: « paralysie flasque rapidement progressive »
Atteinte centrale: rhombencephalite, méningite carcinomateuse, lymphomatose, myélite transverse, compression médullaire Atteinte corne antérieure: poliomyélite, West Nile virus Atteinte racines nerveuses: compression, inflammation, carcinose leptoméningée Atteinte nerf périphérique: porphyrie, diphtérie, M de Lyme, déficit vit B1 (Béri-Béri), vascularite (Lupus), intoxication métaux lourds, drogues, perturbations métaboliques (hypokaliémie, hypophosphatémie, hypermagnésémie, hypoglycémie) Atteinte jonction NM: myasthénie, botulisme, empoisonnement aux organophosphates (pesticides) Atteinte musculaire: polymyosite, dermatomyosite, rhabdomyolyse aigue
, vascularite (Lupus), intoxication métaux lourds, drogues, perturbations métaboliques (hypokaliémie, hypophosphatémie, hypermagnésémie, hypoglycémie) Atteinte jonction NM: myasthénie, botulisme, empoisonnement aux organophosphates (pesticides) Atteinte musculaire: polymyosite, dermatomyosite, rhabdomyolyse aigue.")
38
atteinte respiratoire, troubles déglutition dysautonomie
Sd de Guillain-Barré CAT Risque vital : atteinte respiratoire, troubles déglutition dysautonomie Surveillance régulière +++ (scope) dans ou près de la REA Reanimation si extension rapide tr respiratoires tr rythme card, respi, TA
 dans ou près de la REA. Reanimation si. extension rapide. tr respiratoires. tr rythme card, respi, TA.")
39
Symptomatique +++ : nursing, kiné assistance respiratoire
Sd de Guillain-Barré traitement Symptomatique +++ : nursing, kiné assistance respiratoire sonde gastrique… Spécifique : (ds <2sem) * Immunoglobulines (1g/kg/j, 48h) * plasmaphérèses (2-5échanges)
 * Immunoglobulines (1g/kg/j, 48h) * plasmaphérèses (2-5échanges)")
40
phase d’extension : 12j (1 à 30j) paralysie ascendante
Sd de Guillain-Barré Evolution phase d’extension : 12j (1 à 30j) paralysie ascendante phase en plateau : 11j (1à plusieurs mois) déficit minime, absence de marche (76%) assistance respiratoire (47%) atteinte des paires crâniennes (64%) dysautonomie (64%) tr sphinctériens (16%) phase de récupération : 24sem (1sem à 28 mois) NB: symptomes>2ans pas de récupération complète 40
 paralysie ascendante. phase en plateau : 11j (1à plusieurs mois) déficit minime, absence de marche (76%) assistance respiratoire (47%) atteinte des paires crâniennes (64%) dysautonomie (64%) tr sphinctériens (16%) phase de récupération : 24sem (1sem à 28 mois) NB: symptomes>2ans pas de récupération complète. 40.")
41
séquelles rares chez l’enfant (5 à 10%):
Sd de Guillain-Barré pronostic mortalité < 3% séquelles rares chez l’enfant (5 à 10%): déficit distal, amyotrophie, tremblement ++si symptômes durent >2ans facteurs de mauvais pronostic : * extension rapide * plateau long * atteinte axonale (Campylobacter), atteinte des PC * ventilation assistée 8-16% developperont une CIDP (installation >4-8sem)
: déficit distal, amyotrophie, tremblement. ++si symptômes durent >2ans. facteurs de mauvais pronostic : * extension rapide. * plateau long. * atteinte axonale (Campylobacter), atteinte des PC. * ventilation assistée. 8-16% developperont une CIDP (installation >4-8sem)")
42
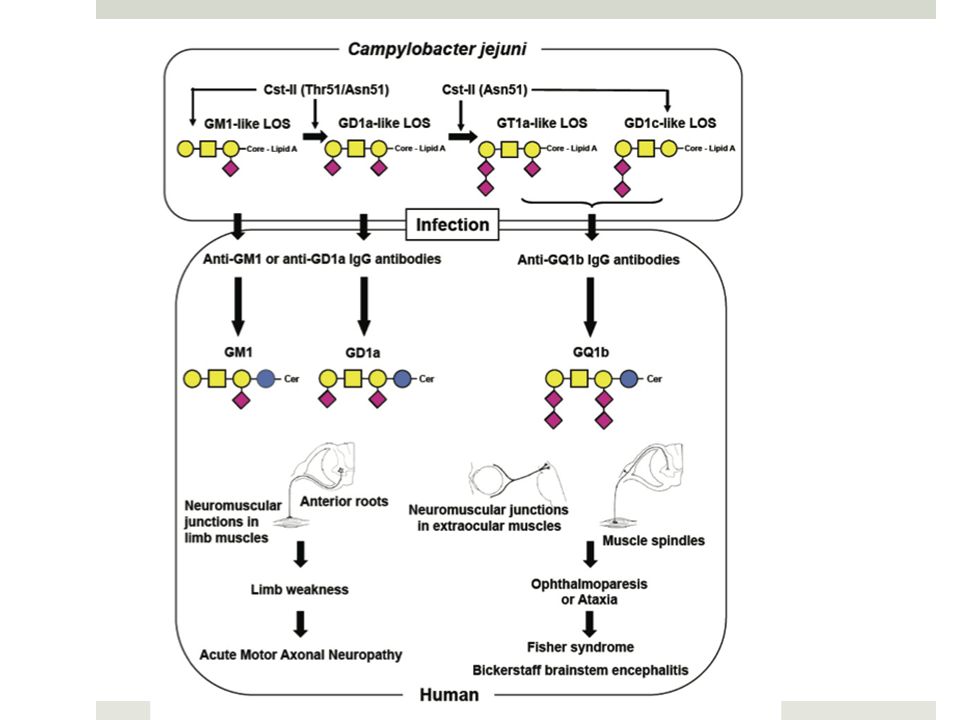
Sd de Guillain-Barré Physiopathologie 1 4 arguments supportent l’hypothèse d’une pathologie auto-immune 50% des patients: AC sériques / gangliosides (GG) (LM1, GM1, GM1b, GM2, GD1a, GalNAc-GD1a, GD1b, GD3, GT1a, GQ1b.). Gangliosides: glycosphingolipides, distribution tissu-spécifique, organisés en micro-domaines fonctionnels = « lipid rafts », role dans la stabilité des membranes cellulaires Réaction croisée, mimétisme moléculaire: C.jejuni isolé de patients GB exprime un lipo-oligosaccharide (LOS) qui imite le groupe carbohydrate d’un type de GG. Le type de GG-like LOS est codé génétiquement. Modèle animal: immunisation avec GM1-like LOS (issus de patients) => tab de NP axonale = tab des patients GG= large famille de glycosphingolipides Guérison des patients s’accompagnait d’une chute des anti-GG: 1988, Ilyas et al
 (LM1, GM1, GM1b, GM2, GD1a, GalNAc-GD1a, GD1b, GD3, GT1a, GQ1b.). Gangliosides: glycosphingolipides, distribution tissu-spécifique, organisés en micro-domaines fonctionnels = « lipid rafts », role dans la stabilité des membranes cellulaires. Réaction croisée, mimétisme moléculaire: C.jejuni isolé de patients GB exprime un lipo-oligosaccharide (LOS) qui imite le groupe carbohydrate d’un type de GG. Le type de GG-like LOS est codé génétiquement. Modèle animal: immunisation avec GM1-like LOS (issus de patients) => tab de NP axonale = tab des patients. GG= large famille de glycosphingolipides. Guérison des patients s’accompagnait d’une chute des anti-GG: 1988, Ilyas et al.")
44
Sd de Guillain-Barré Physiopathologie 2 Études post-mortem => présence de complément activé (complexe d’attaque membranaire) au niveau des lésions nerveuses: cellules de schwann (AIDP) et axolemme (AMAN) anti-GM1 affectent les canaux sodiques des nœuds de Ranvier (modèle lapin) Facteurs prédisposants (SNP?) pourraient influencer le suceptibilité au GB => Perspectives thérapeutiques! To summarise, in AMAN subsequent to C Jejuni enteritis, infection by C jejuni bearing the GM1-like LOS can induce the production of IgG anti-GM1 antibodies. The autoantibodies bind to GM1 at the nodes of Ranvier in the spinal anterior roots, and activate complement. Membrane attack complex is formed at the nodal axolemma, which leads to “disappearance” of sodium channel clusters and disruption of axo-glial junctions. The pathological changes are able to produce muscle weakness. In severe cases, axonal degeneration occurs subsequently.
 au niveau des lésions nerveuses: cellules de schwann (AIDP) et axolemme (AMAN) anti-GM1 affectent les canaux sodiques des nœuds de Ranvier (modèle lapin) Facteurs prédisposants (SNP ) pourraient influencer le suceptibilité au GB. => Perspectives thérapeutiques! To summarise, in AMAN subsequent to C Jejuni enteritis, infection by C jejuni bearing the GM1-like LOS can induce the production of IgG anti-GM1 antibodies. The autoantibodies bind to GM1 at the nodes of Ranvier in the spinal anterior roots, and activate complement. Membrane attack complex is formed at the nodal axolemma, which leads to disappearance of sodium channel clusters and disruption of axo-glial junctions. The pathological changes are able to produce muscle weakness. In severe cases, axonal degeneration occurs subsequently.")
45
Merci LEUKO FRANCE Centre de référence Maladies Rares
« leucodystrophies »

Présentations similaires








. Il s’agit ici de nerfs rachidiens. A distinguer.>")




>")



 C. Mouala 1,2, S. Houzé 3,>")
>")

