Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Réglementation du médicament
IFSI Bourges 3A Réglementation du médicament Introduction générale Philippe Costes

2
I. Définition Article L.511 du Code de la Santé Publique :
« On entend par médicament toute substance ou composition présentée comme possédant des propriétés curatives ou préventives à l’égard des maladies humaines ou animales, ainsi que tout produit pouvant être administré à l’homme ou à l’animal, en vue d’établir un diagnostic médical ou de restaurer, corriger ou modifier leurs fonctions organiques ».

3
I. Définition Le médicament sert : À soigner, ex. antibiotique
À prévenir, ex. vaccin À diagnostiquer, ex. produit de contraste À restaurer, corriger, modifier les fonctions de l’organisme, ex. vitamine Les produits vétérinaires sont des médicaments Sont aussi considérés comme médicaments les produits cosmétiques et diététiques renfermant des substances ou produits entrant dans cette définition.

4
I. Définition Chaque médicament contient un PRINCIPE ACTIF
(= PA) responsable de l’action thérapeutique. Ce PA existe sous trois noms: Le nom chimique La DCI = Dénomination Commune Internationale Le nom commercial, nom déposé par le laboratoire qui exploite le PA sous forme d’une spécialité ex. paracétamol: doliprane®
 responsable de l’action thérapeutique. Ce PA existe sous trois noms: Le nom chimique. La DCI = Dénomination Commune Internationale. Le nom commercial, nom déposé par le laboratoire qui exploite le PA sous forme d’une spécialité. ex. paracétamol: doliprane®")
5
I. Définition Les différentes catégories de médicaments :
Les préparations magistrales: médicaments préparés extemporanément en pharmacie selon une prescription médicale pour un patient donné. Les préparations hospitalières ou officinales: préparation effectuées en pharmacie selon les indications de la Pharmacopée en l’absence de spécialités existante ou adaptée, et destinées à un ou plusieurs malades Les spécialités pharmaceutiques: mdts préparés à l’avance par l’industrie pharmaceutique sous un conditionnement, et caractérisées par le nom commercial.

8
Cas particulier : Le Générique
Art. L (1996) du Code de la SP : « on entend par spécialité générique d'une autre spécialité, une spécialité qui a : -la même composition qualitative et quantitative en principes actifs, -la même forme pharmaceutique, -et dont la bioéquivalence avec la spécialité de référence a été démontrée par des études appropriées de biodisponibilité."
 du Code de la SP : « on entend par spécialité générique d une autre spécialité, une spécialité qui a : -la même composition qualitative et quantitative en principes actifs, -la même forme pharmaceutique, -et dont la bioéquivalence avec la spécialité de référence a été démontrée par des études appropriées de biodisponibilité.")
9
Un médicament générique est un médicament identique ou équivalent à celui d'une marque (appelé médicament princeps), mais produit et vendu sous sa DCI. Le dossier requis par l’AFSSAPS pour l'enregistrement de cette copie est plus simple par rapport à celui du produit original (dispense d'études pharmaco-toxico-cliniques). Ces médicaments génériques sont généralement produits après expiration du brevet du médicament princeps. Un médicament générique est généralement vendu à un prix moindre. Toutefois, certains laboratoires ont décidé de baisser le prix de leur médicaments princeps pour encourager les médecins à continuer à les prescrire.
. Ces médicaments génériques sont généralement produits après expiration du brevet du médicament princeps. Un médicament générique est généralement vendu à un prix moindre. Toutefois, certains laboratoires ont décidé de baisser le prix de leur médicaments princeps pour encourager les médecins à continuer à les prescrire.")
10
II. Le Monopole Pharmaceutique
Terme qui signifie « le seul à vendre » L’article L512 du CSP réserve aux pharmaciens la préparation et la délivrance : - des médicaments, - des objets de pansements, - de tous les articles présentés comme conformes à la pharmacopée.

11
II. Le Monopole Pharmaceutique
A l’hôpital, c’est la loi du 8/12/1992 qui reconnaît l’existence des pharmacies à usage intérieur (PUI). L’article L592-2 définit leurs missions, notamment : la gestion, l’approvisionnement, la préparation, le contrôle, la détention , la dispensation des mdts, des produits ou objets mentionnés à l’article L512, et des dispositifs médicaux stériles.
. L’article L592-2 définit leurs missions, notamment : la gestion, l’approvisionnement, la préparation, le contrôle, la détention , la dispensation des mdts, des produits ou objets mentionnés à l’article L512, et des dispositifs médicaux stériles.")
12
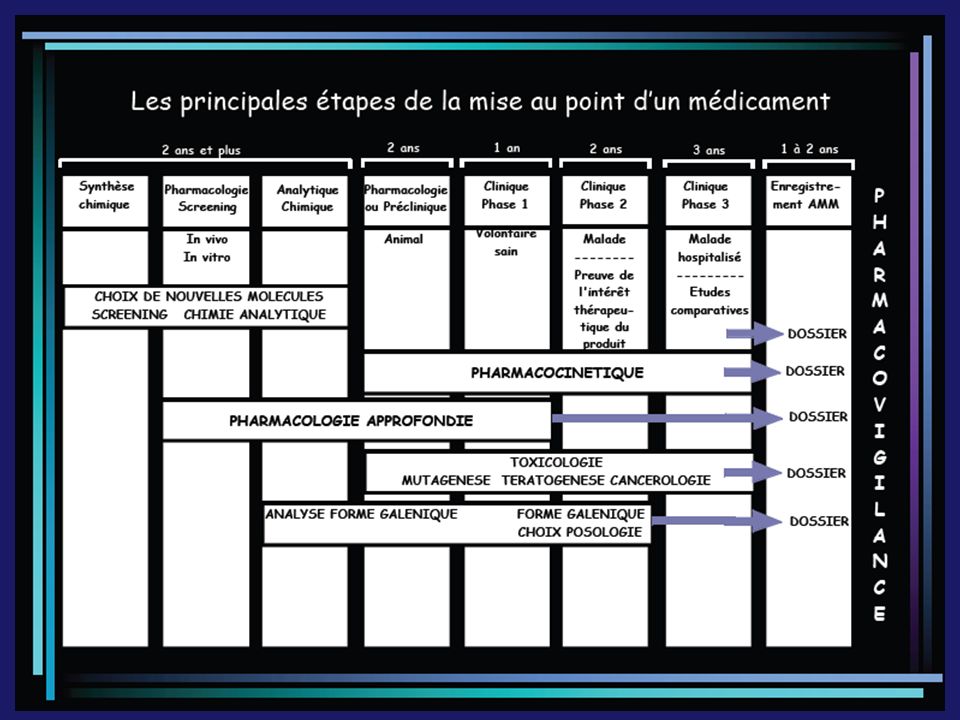
III. Le Développement d’un mdt A. Essais cliniques
Décret du 26 avril 2006: règle le déroulement des essais cliniques afin de protéger les personnes qui se prêtent à des recherches biomédicales et de garantir la rigueur scientifique de ces études. Ces essais sont mis en place par un promoteur (généralement le laboratoire pharmaceutique) auprès d’investigateurs (médecins, pharmaciens) Ces essais ne peuvent débuter qu’après un avis favorable du CPP = Comite pour la Protection des Personnes
 auprès d’investigateurs (médecins, pharmaciens) Ces essais ne peuvent débuter qu’après un avis favorable du CPP = Comite pour la Protection des Personnes.")
13
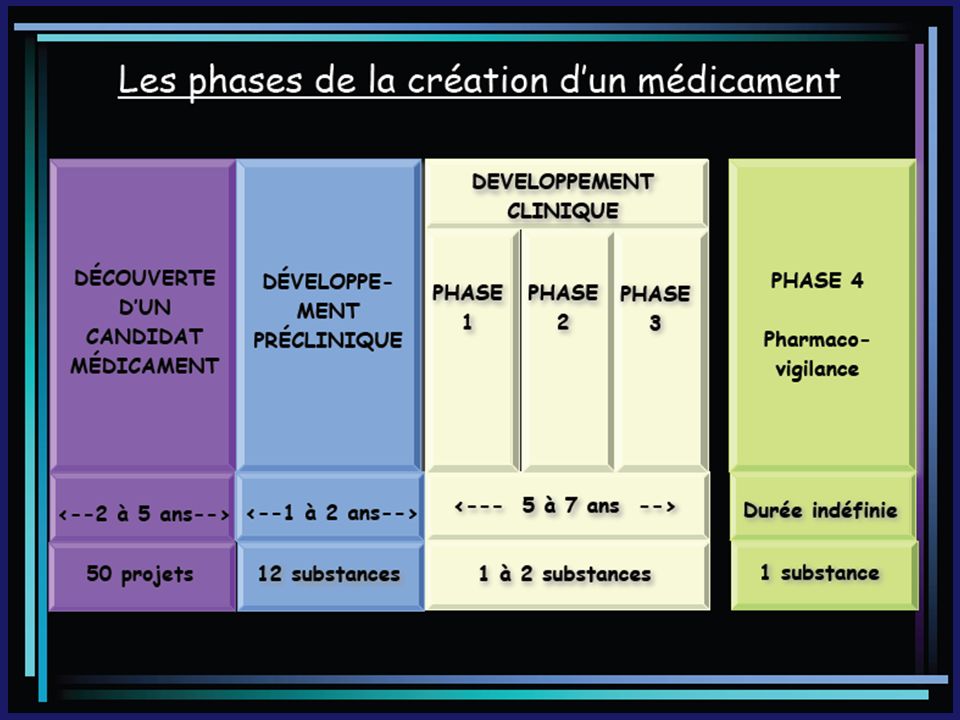
III. Le Développement d’un mdt A. Essais cliniques
Dans le cadre de l’étude d’un nouveau médicament, les études cliniques se répartissent en 4 phases

15
III. Le Développement d’un mdt A. Essais cliniques
Dans le cadre de l’étude d’un nouveau médicament, les études cliniques se répartissent en 4 phases: Phase I: premières administrations chez des volontaires sains (petits groupes) afin de déterminer les doses à administrer et les effets indésirables potentiellement dangereux. Les études pharmacocinétique et de métabolisme font également partie de cette phase.
 afin de déterminer les doses à administrer et les effets indésirables potentiellement dangereux. Les études pharmacocinétique et de métabolisme font également partie de cette phase.")
16
III. Le Développement d’un mdt A. Essais cliniques
Phase II: mise en évidence de l’activité thérapeutique au cours d’essais de courte durée, portant sur de petits effectifs de malades aussi homogènes que possible Phase III: démonstration de l’activité thérapeutique au cours d’essais de durée prolongée, portants sur des effectifs importants de malades et comparaison aux traitements de référence

17
III. Le Développement d’un mdt A. Essais cliniques
Phase IV: ces essais ont lieu après commercialisation du mdt. Ils ont pour but de détecter les effets indésirables rares mais graves non détectés sur de courtes durées. Ils permettent aussi d’obtenir des extensions d’AMM soit pour de nouvelles indications soit pour de nouvelles catégories de patients.

18
III. Le Développement d’un mdt B. Autorisation de mise sur le marché
L’autorisation de mise sur le marché ou AMM est obligatoire pour la commercialisation d’un nouveau mdt. Elle est délivrée par l’AFSSAPS = Agence Française de Sécurité Sanitaire des Aliments et des Produits de Santé (ex. agence du médicament)
")
19
III. Le Développement d’un mdt B. Autorisation de mise sur le marché
Le dossier d’AMM est constitué de 4 parties: Résumé du dossier (administratif, résumé des caractéristiques du produit = RCP) Documentation chimique, pharmaceutique, biologique Documentation toxicologique et pharmacologique (toxicité aiguë et chronique, tests sur la reproduction, effet mutagène). Etudes effectuées chez l’animal Documentation clinique, pharmacologie humaine et résultats des essais cliniques
 Documentation chimique, pharmaceutique, biologique. Documentation toxicologique et pharmacologique (toxicité aiguë et chronique, tests sur la reproduction, effet mutagène). Etudes effectuées chez l’animal. Documentation clinique, pharmacologie humaine et résultats des essais cliniques.")
20
III. Le Développement d’un mdt B. Autorisation de mise sur le marché
La commission d’AMM évalue, après avis de rapporteurs, la qualité, l’efficacité, la tolérance et le rapport bénéfice / risque du nouveau médicament. L’AMM est délivrée pour 5 ans, avec renouvellement par période quinquennale. Elle est accompagnée d’un n° d’AMM et du RCP, qui a pu être modifié par l’Agence. Il existe une Agence Européenne pour l’Évaluation du Médicament, qui permet aux labo. d’obtenir une AMM européenne.

21
III. Le Développement d’un mdt C. La commission de transparence
Après l’obtention de l’AMM, c’est la Commission de Transparence qui émet un avis sur l’Amélioration du Service Médical Rendu (ASMR) par le nouveau médicament en regard des médicaments existants. Echelle de 1 à 6: 1 = amélioration majeure 6 = avis défavorable La commission de transparence élabore des fiches de transparence afin de promouvoir le bon usage du mdt et d’éviter les dépenses injustifiées.
 par le nouveau médicament en regard des médicaments existants. Echelle de 1 à 6: 1 = amélioration majeure. 6 = avis défavorable. La commission de transparence élabore des fiches de transparence afin de promouvoir le bon usage du mdt et d’éviter les dépenses injustifiées.")
22
III. Le Développement d’un mdt C. La commission de transparence
Elle propose un taux de remboursement par la Sécurité Sociale, à savoir: 100% = spécialités pharmaceutiques reconnues comme irremplaçables et/ou particulièrement coûteuses. Vignette blanche, marquée d’une croix 65% = ASMR en terme d’efficacité ou d’effet indésirable, ou économie dans le coût d’un traitement médicamenteux Vignette blanche 35% = mdts destinés aux troubles et affections sans gravité habituelle (« de confort ») - mdts dont le service médical n'a pas été reconnu comme majeur. Vignette bleue
 - mdts dont le service médical n a pas été reconnu comme majeur. Vignette bleue.")
23
III. Le Développement d’un mdt C. La commission de transparence
NB: un laboratoire peut ne pas demander de remboursement par la sécurité sociale. Dans ce cas, le mdt n’est pas remboursé et son prix est libre. Mdt « conseil » ou « grand public » Seules les spécialités « agrées aux collectivités » peuvent être achetées et utilisées par les établissements de soins. C’est la commission de transparence qui en fixe la liste.

24
III. Le Développement d’un mdt D. Le Prix
Une commission spécifique va définir un prix de vente du médicament en tenant compte des efforts de recherche du laboratoire et du marché potentiel (forte négociation avec le laboratoire). Tous ces avis (AMM, remboursement, prix, agrément aux collectivités) font l’objet d’une parution au JO
. Tous ces avis (AMM, remboursement, prix, agrément aux collectivités) font l’objet d’une parution au JO.")
26
IV. Les autres statuts du mdt ATU
ATU = Autorisation Temporaire d’Utilisation. Procédure exceptionnelle permettant de mettre à disposition des patients, des mdts qui n’ont pas d’AMM. Les ATU sont accordées par l’AFSSAPS.

27
V. Les Substances vénéneuses
Les médicaments doivent leur activité à des substances qui, en fonction de la dose utilisée, peuvent se révéler vénéneuses. La réglementation relative à la délivrance des médicaments a pour but d'apporter à l'utilisateur les garanties maximales de sécurité dans l'emploi des médicaments. Elle impose, pour cela, des restrictions à la délivrance, à l’étiquetage et au stockage des médicaments contenant des substances vénéneuses. Ces médicaments sont classés en "liste".

28
V. Les Substances vénéneuses A. Classification
Les substances vénéneuses sont inscrites sur 3 listes: Liste I: mdt toxique Liste II: mdt dangereux Stupéfiants: mdt toxicomanogène La délivrance médicaments contenant des substances vénéneuses ne peut se faire que sur prescription médicale Les mdts non inscrits sur une liste peuvent être vendus sans ordonnance en officine.

29
V. Les Substances vénéneuses B. Étiquetage
Sont obligatoires pour toutes les spécialités: Conditionnement primaire: Nom commercial, DCI, forme, dosage, labo., n° de lot, date de péremption, n°AMM Emballage externe: idem + nombre de formes unitaires, adresse labo., conditions de conservation, vignette pour le remboursement... Notice d’utilisation: RCP.

30
V. Les Substances vénéneuses B. Etiquetage
Pour les substances vénéneuses, l’emballage externe et le conditionnement primaire doivent présenter: Liste I et Stupéfiants: un liseré rouge Liste II: un liseré vert Deux mentions sont inscrites sous ces cadres: « Respecter les doses prescrites » « ne peut être obtenu que sur prescription médicale » Éventuellement: « Ne pas avaler »

31
V. Les Substances vénéneuses C. Règles de prescription
Ordonnance obligatoire pour les médicaments inscrits sur une liste. (et pour obtenir le remboursement sécu) Qui prescrit?: En ville: les titulaires du diplôme d’état de Docteur en médecine A l ’hôpital Sans limitation: PH, assistants spécialistes ou généralistes, attachés Avec limitation: chirurgiens dentistes, sages femmes Par délégation du médecin dont ils relèvent: internes, résidents (mais interdiction pour les stupéfiants)
 Qui prescrit : En ville: les titulaires du diplôme d’état de Docteur en médecine. A l ’hôpital. Sans limitation: PH, assistants spécialistes ou généralistes, attachés. Avec limitation: chirurgiens dentistes, sages femmes. Par délégation du médecin dont ils relèvent: internes, résidents (mais interdiction pour les stupéfiants)")
32
V. Les Substances vénéneuses C. Règles de prescription
Mentions obligatoires sur l’ordonnance: Identification du prescripteur Adresse du prescripteur (ou de l’établissement) Identification du patient: nom, prénom, sexe, âge, poids et taille (enfants) Identification du ou des mdts: nom, forme, dosage, posologie, durée d’administration, voie d’administration Date et signature du prescripteur ALD = Affection Longue Durée (liste au JO): prescription sur une ordonnance « bi-zone ». Prise en charge à 100%.
 Identification du patient: nom, prénom, sexe, âge, poids et taille (enfants) Identification du ou des mdts: nom, forme, dosage, posologie, durée d’administration, voie d’administration. Date et signature du prescripteur. ALD = Affection Longue Durée (liste au JO): prescription sur une ordonnance « bi-zone ». Prise en charge à 100%.")
33
V. Les Substances vénéneuses D. Règles de délivrance
Liste II Liste I Stupéfiants Délivrance au public Ordonnance < 3 mois idem Ordonnance sécurisée Quantité dispensée 1 mois Règle des 7, 14 ou 28 jrs Durée de validité de prescription 12 mois 12 mois sauf cas particuliers 7, 14 ou 28 jrs Renouvellement Autorisé sauf si le prescripteur l'a interdit Autorisé seulement si le prescripteur l'a expressément indiqué INTERDIT Stockage Lieu clos hors de portée du public Coffre

34
VI. Les mdts à prescription restreinte A
VI. Les mdts à prescription restreinte A. Mdt réservé à l’usage hospitalier Classement justifié soit: Par caractéristiques pharmacologiques du mdt Par son caractère innovant Pour des motifs de santé publique Prescripteur = praticien exerçant dans un établissement de santé publique ou privé Dispensation effectuée par une PUI. Rétrocession si patient ambulatoire.

35
Rétrocession La rétrocession hospitalière est la dispensation par une pharmacie hospitalière de médicaments, achetés par l'hôpital, à des patients ambulatoires. Les dépenses de médicaments rétrocédés ne sont pas imputées sur le budget de l'hôpital mais présentées au remboursement en soins de ville au titre de ces patients non hospitalisés.

36
VI. Les mdts à prescription restreinte B
VI. Les mdts à prescription restreinte B. Mdt à prescription initiale hospitalière Classement justifié soit: Par le diagnostic de la pathologie à traiter Par les caractéristiques pharmacologiques du mdt Pour des motifs de santé publique La prescription initiale doit être effectuée par un praticien hospitalier, le renouvellement pouvant être effectué par un médecin généraliste Dispensation en officine de ville

37
VI. Les mdts à prescription restreinte C
VI. Les mdts à prescription restreinte C. Mdt nécessitant une surveillance particulière pendant le ttt Classement justifié par la gravité des effets indésirables pouvant survenir au cours du ttt. Ex.: méthadone

38
VII. Les mdts d’exception
Ce classement est justifié par l’existence de spécialités coûteuses et d’indications précises. Une «fiche d’information thérapeutique» est rédigée par la commission de transparence afin d’assurer une bonne utilisation de chaque spécialité. La prescription doit être effectuée sur une ordonnance à 4 volets de mdts d’exception. Le prescripteur s’engage au respect strict de la fiche d’information. Dispensation en officine de ville.

Présentations similaires










>")










