Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Mécanismes des maladies héréditaires
Anomalies du génome Mécanismes des maladies héréditaires I – Réarrangements ou Mutations de grande taille II - Mutations de petite taille III – Mutations dynamiques : expansions de triplets IV - Epimutations

3
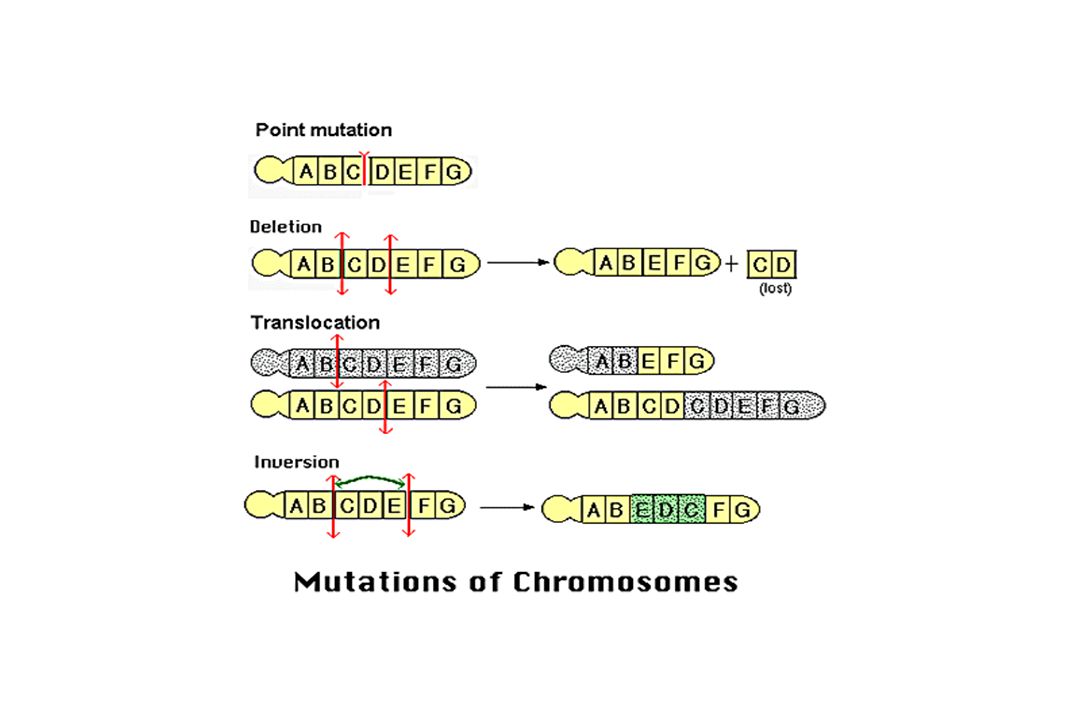
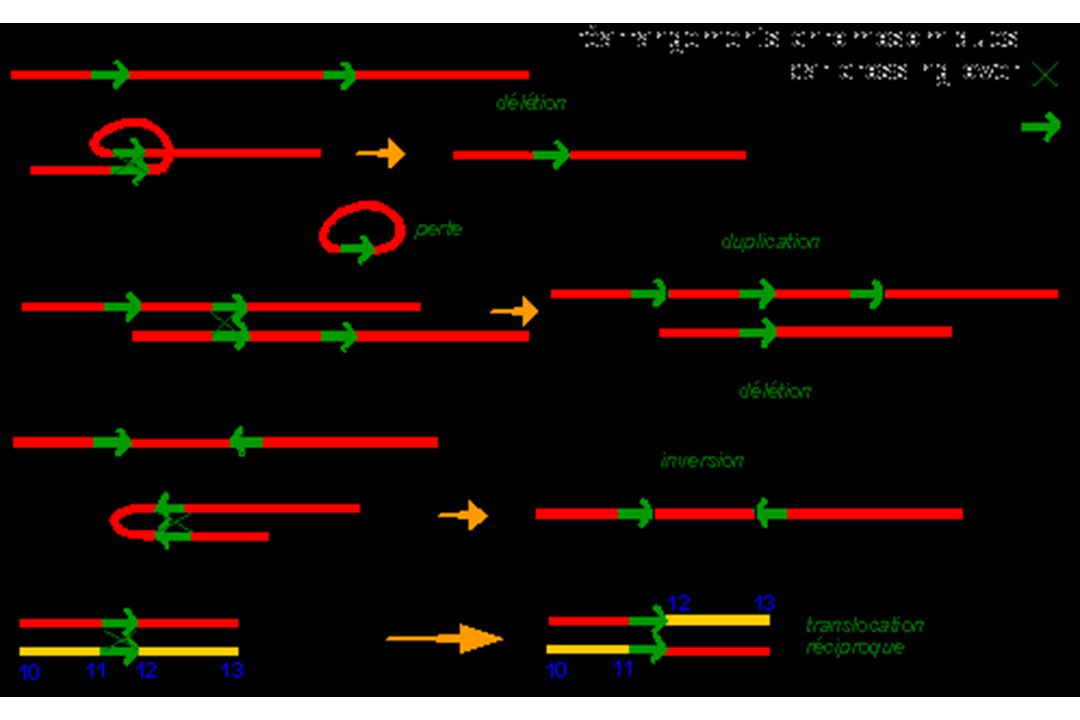
I – Réarrangements de grande taille
A) Délétion B) Duplication C) Inversion D) Conversion E) Insertion
 Délétion. B) Duplication. C) Inversion. D) Conversion. E) Insertion.")
6
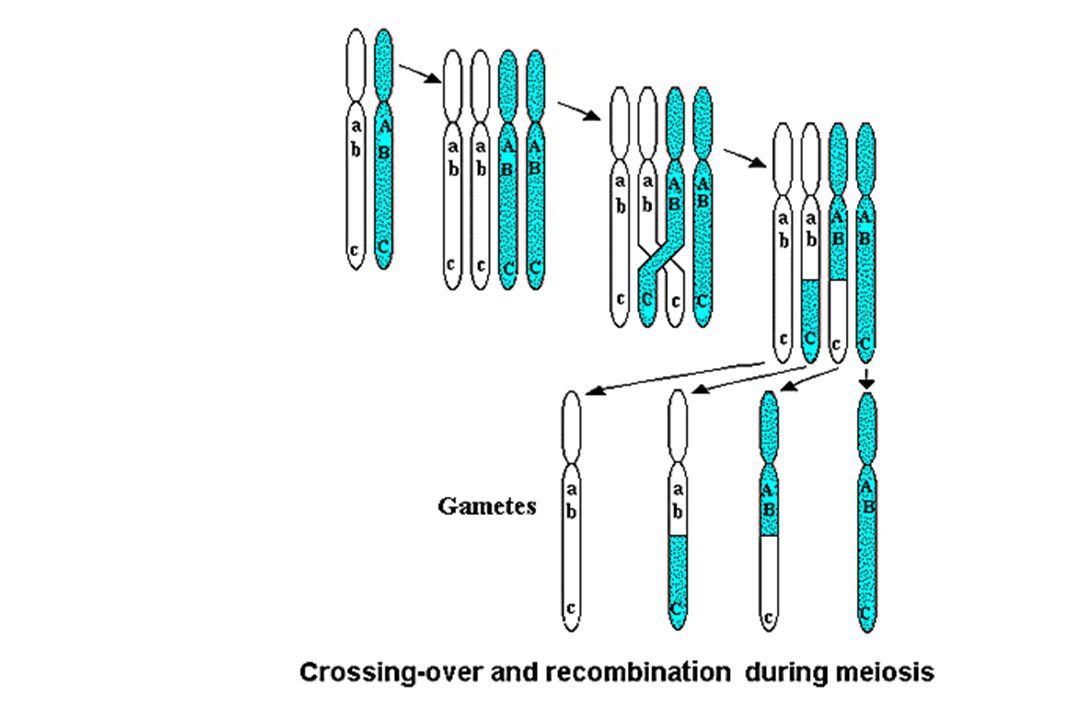
Recombinaisons égales - 30 à 40 par méïose
- échanges d’allèles : variabilité génétique - les recombinaisons sont plus fréquentes entre gènes éloignés - estimation de la distance génétique entre deux gènes exprimée en centimorgans cM 1 cM = 1000Kb Recombinaisons inégales - entraîne des délétions - entraîne des insertions ou duplications A B A B A B A B

7
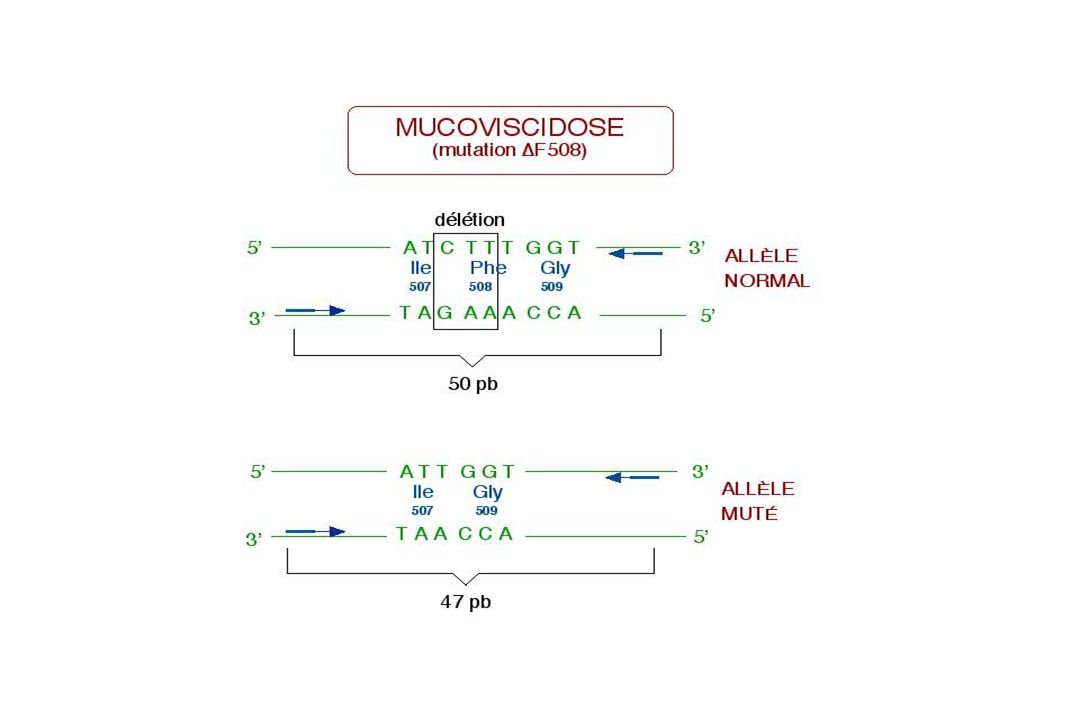
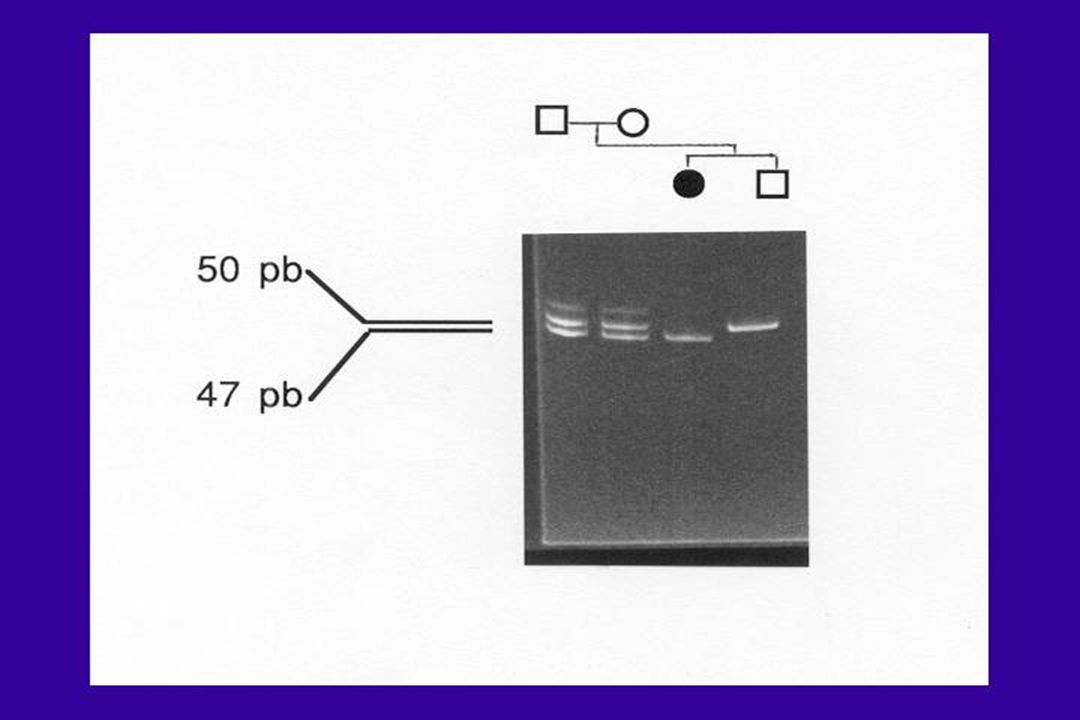
Délétions - Duplications
Souvent observé au sein de familles de gènes regroupés en groupes = « clusters » Exemple : gènes de la famille -globine (16p13.3) et -thalassémies. Phénomènes peu fréquents = 5% Sauf Chromosome X 5% à 95% Ex : gène de la dystrophine (myopathie de Duchenne) CNV : copy number variation
 et. -thalassémies. Phénomènes peu fréquents = 5% Sauf Chromosome X 5% à 95% Ex : gène de la dystrophine (myopathie de Duchenne) CNV : copy number variation.")
10
C) Inversion Changement d’orientation d’un segment d’ADN.
 Inversion Changement d’orientation d’un segment d’ADN.")
11
Transfert unidirectionnel d’information génétique
D) Conversion génique Transfert unidirectionnel d’information génétique d’un chromosome à l’autre A B récepteur A B donneur A B A A B A B Exemples : Maladie de Gaucher de type I, hyperplasie congénitale des surrénales
 Conversion génique. Transfert unidirectionnel d’information génétique. d’un chromosome à l’autre. A. B. récepteur. A. B. donneur. A. B. A. A. B. A. B. Exemples : Maladie de Gaucher de type I, hyperplasie. congénitale des surrénales.")
13
E) Insertion Introduction d’une séquence (transposon ou séquence virale) dans un gène. La mutation par insertion résulte de plusieurs mécanismes complexes (recombinaison inégale, translocation, conversion génique, « boucles chromatiniennes »). Séquences courtes SINE Alu gène NF1 : neurofibromatose Séquences longues LINE L1 transposon gène Facteur VIII : hémophilie
. Séquences courtes SINE Alu. gène NF1 : neurofibromatose. Séquences longues LINE L1 transposon. gène Facteur VIII : hémophilie.")
14
II – MUTATIONS DE PETITE TAILLE
- Modification de l’ADN sous l’effet d’agents physiques ou chimiques – absence de réparation - Erreurs de fidélité de l’ADN polymérase lors de la replication - Dépurination Désamination notam. des cytosines méthylées (metC T sur le brin sens et G A sur l’antisens) Transition : purine purine pyrimidine pyrimidine Transversion : purine pyrimidine
 Transition : purine purine. pyrimidine pyrimidine. Transversion : purine pyrimidine.")
16
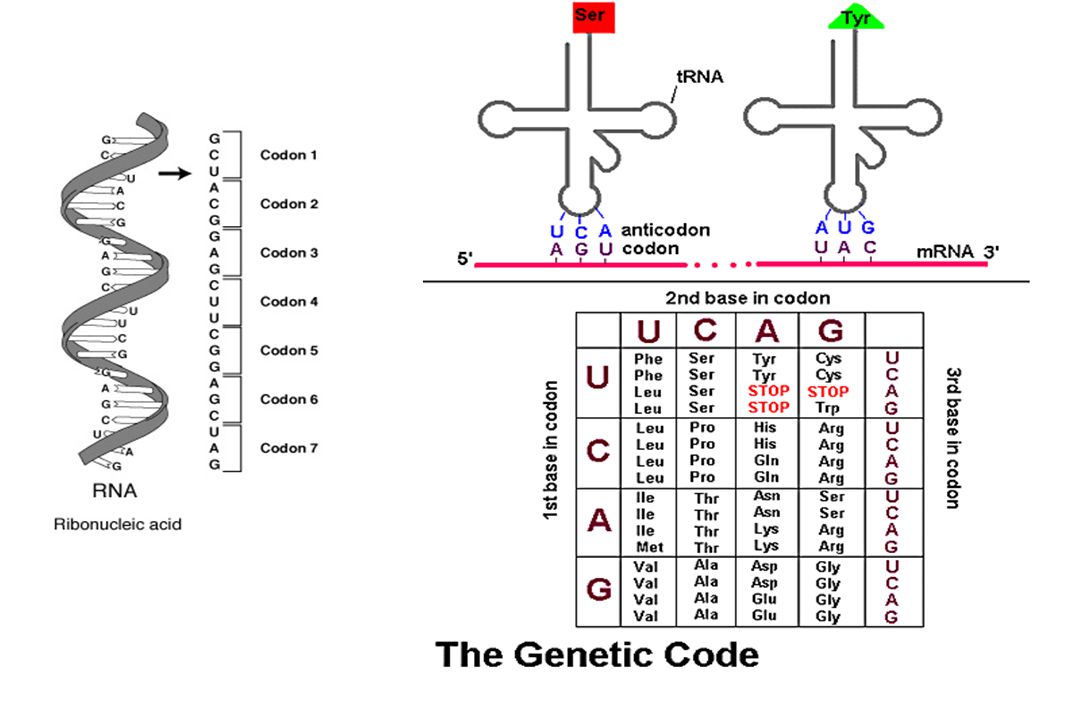
I 1 – Mutations ponctuelles
Neutre hors séquence codante AA AA1 Faux-sens AA AA2 Non-sens ou Stop AA STOP (TAA, TAG, TGA)
")
17
MUTATION FAUX -SENS p.Glu6Val

18
MUTATION STOP ou non-sens
Nomenclature G542X p.Gly542X

19
MUTATION DANS UN CODON STOP
UAA/UAG/UGA codon avec sens Allongement de la chaîne peptidique

20
I 1– Mutations ponctuelles
Neutre AA AA1 Faux-sens AA AA2 Non-sens ou Stop AA STOP I2 – Délétions insertion d’un nucléotide Décalage du cadre de lecture

21
ARN obtenu à partir d’un ADN normal
ARN obtenu à partir d’un ADN présentant une insertion

23
I 1– Mutations ponctuelles
Neutre AA AA1 Faux-sens AA AA2 Non-sens ou Stop AA STOP I2 – Délétions insertion d’un nucléotide I3 – Délétion d’un codon

26
I 1– Mutations ponctuelles
Neutre AA AA1 Faux-sens AA AA2 Non-sens ou Stop AA STOP I 2 – Délétions insertion d’un nucléotide I 3 – Délétion d’un codon I 4 – Mutations au niveau d’un site d’épissage

27
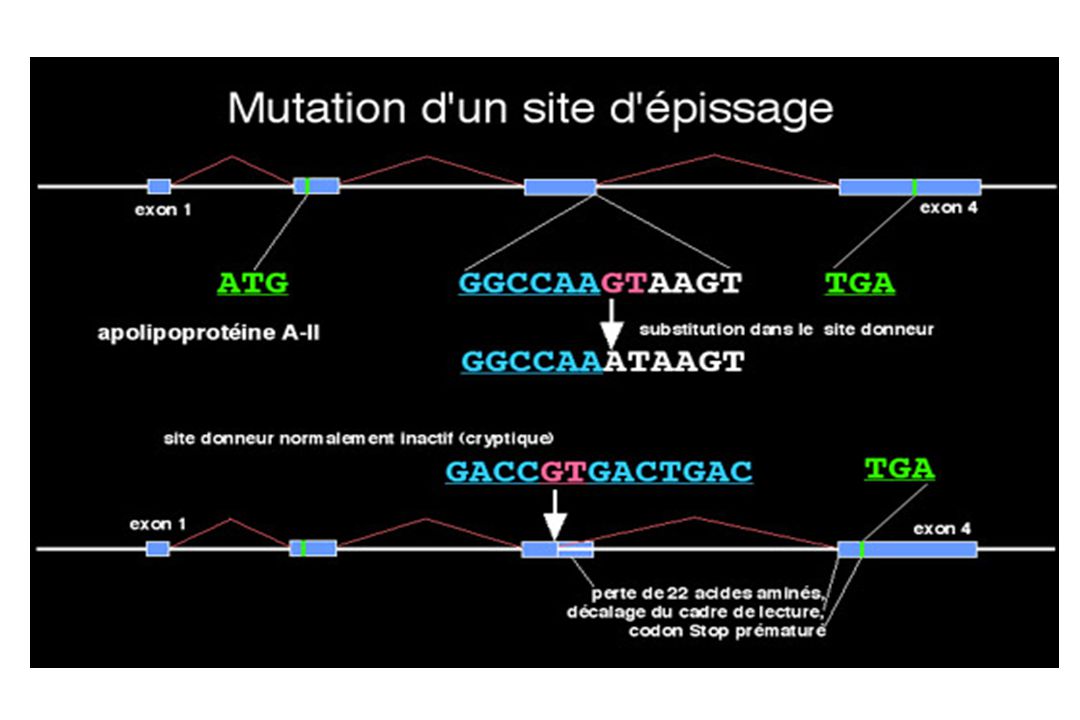
I 4 – Mutations au niveau d’un site d’épissage
EXON EXON INTRON 5’ GTT CTT GGA GAA GGT GTACTTGGATCCTGAAAG GAG GTG AAG 3’ val leu gly glu gly glu val lys GT : site donneur AG : site accepteur 5’ GTT CTT GGA GAA GGT TTA CTT GGA TCC TGA AAG GAG GTG AAG 3’ val leu gly glu gly leu leu gly ser stop lys glu val lys

29
- au niveau d’un site d’épissage
- au niveau des séquences ISE (intronic splicing enhancer) et ESE (exonic splicing enhancer) = 15% des anomalies responsables de maladies génétiques - saut d’exon (exon skipping) protéine tronquée (50% des cas) - activation d’un site cryptique d’épissage - création d’un « pseudo-exon » dans un intron - rétention d’un intron dans l’ARNm mature
 et ESE (exonic splicing enhancer) = 15% des anomalies responsables de maladies génétiques. - saut d’exon (exon skipping) protéine tronquée (50% des cas) - activation d’un site cryptique d’épissage. - création d’un « pseudo-exon » dans un intron. - rétention d’un intron dans l’ARNm mature.")
30
I – Réarrangements ou Mutations de grande taille
II - Mutations de petite taille III – Mutations dynamiques : expansions de triplets IV - Epimutations

31
III – Amplifications ou expansions de triplets
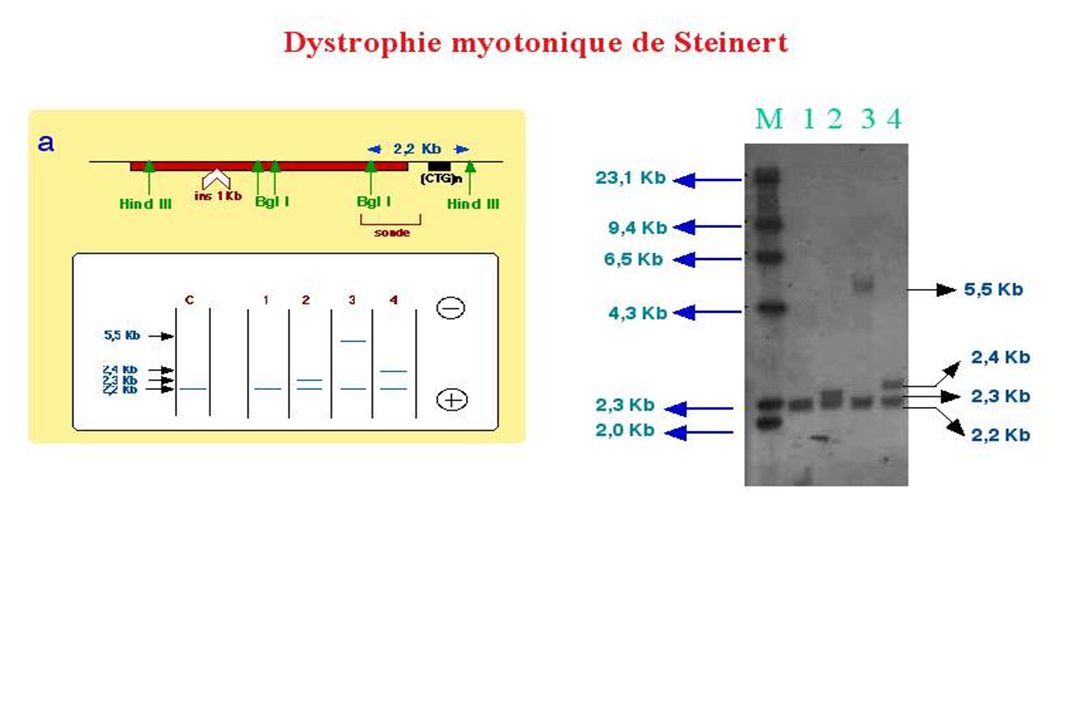
Maladie Nature du triplet Nombre de normal triplets pathologique Sd X fragile (CGG)n n=6-54 N > 200 Sd de Kennedy (CAG)n N=14-32 N > 40 Dystrophie myotonique de Steinert (CTG)n N=5-37 N > 50 Chorée de Huntington N=6-35 N > 37 SCA1 N=6-39 Retard mental FRAXE N=4-39 Sd de Jacobson N=11 N > 100 DRPLA N=3-36 N > 49 SCA3 N=13-44 N > 60 Ataxie de Friedreich (GAA)n N=6-29 SCA7 N=7-35 SCA2 N=13-32 N > 33 SCA6 N=4-18 N > 21
n. n=6-54. N > 200. Sd de Kennedy. (CAG)n. N= N > 40. Dystrophie myotonique. de Steinert. (CTG)n. N=5-37. N > 50. Chorée de Huntington. N=6-35. N > 37. SCA1. N=6-39. Retard mental FRAXE. N=4-39. Sd de Jacobson. N=11. N > 100. DRPLA. N=3-36. N > 49. SCA3. N= N > 60. Ataxie de Friedreich. (GAA)n. N=6-29. SCA7. N=7-35. SCA2. N= N > 33. SCA6. N=4-18. N > 21.")
32
Classification des maladies à amplification de triplets
A- Les maladies à expansion de triplets non codants expansions de grande taille instabilité élevée phénotype : atteinte multisystémique de nombreux organes les manifestations cliniques sont variables dans une même famille du fait d’une hétérogénéité somatique la nature de la séquence répétée est variable suivant les pathologies CGG, CTG, CAA les mécanismes pathogéniques diffèrent selon les pathologies et dépendent des conséquences de la perte de fonction du produit du gène

33
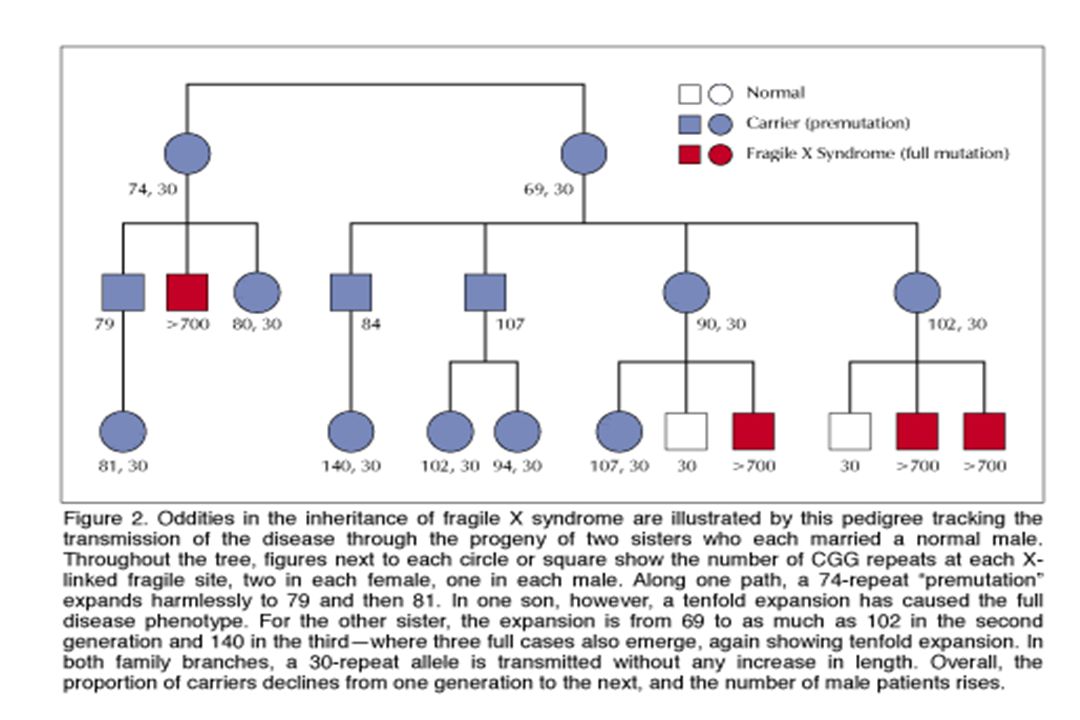
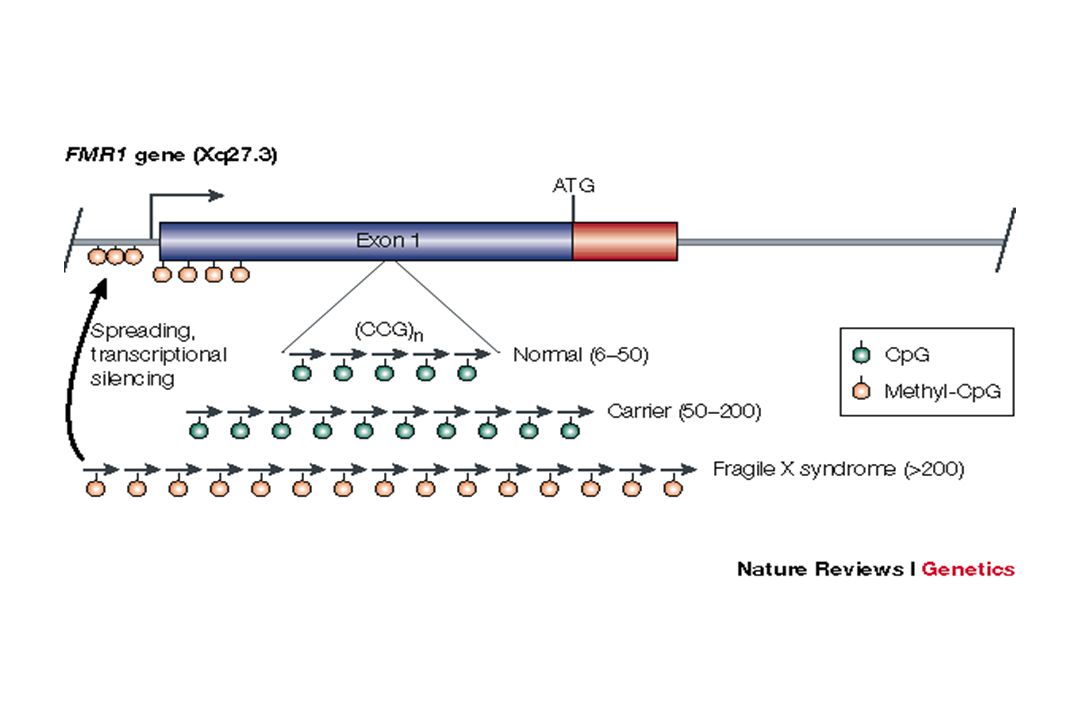
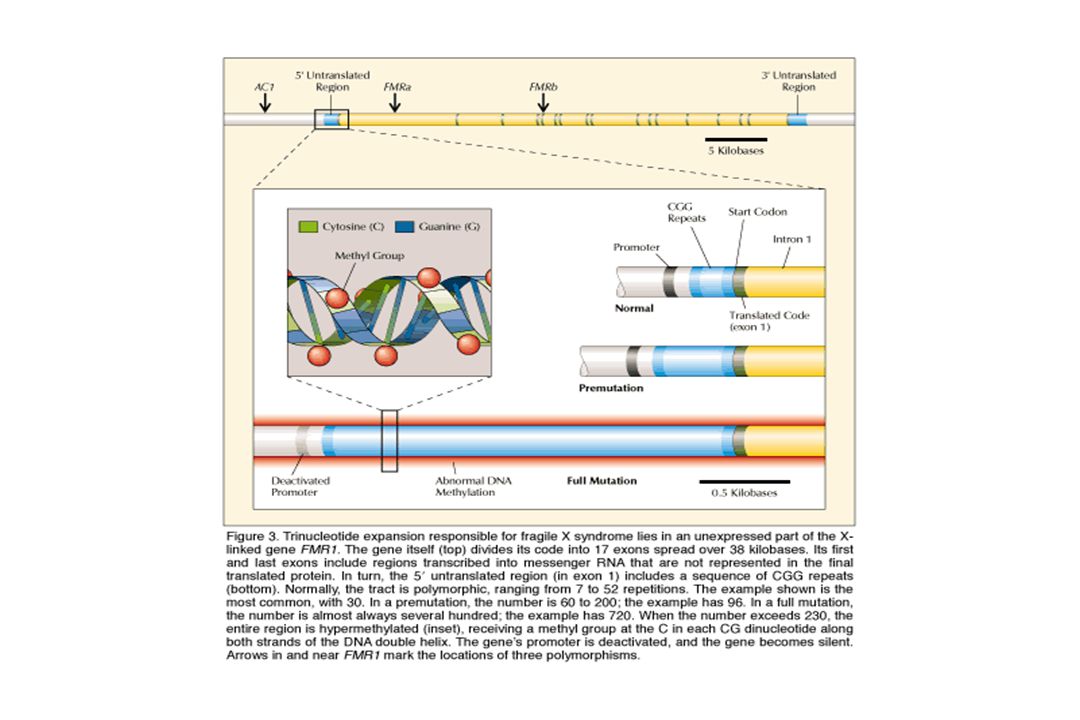
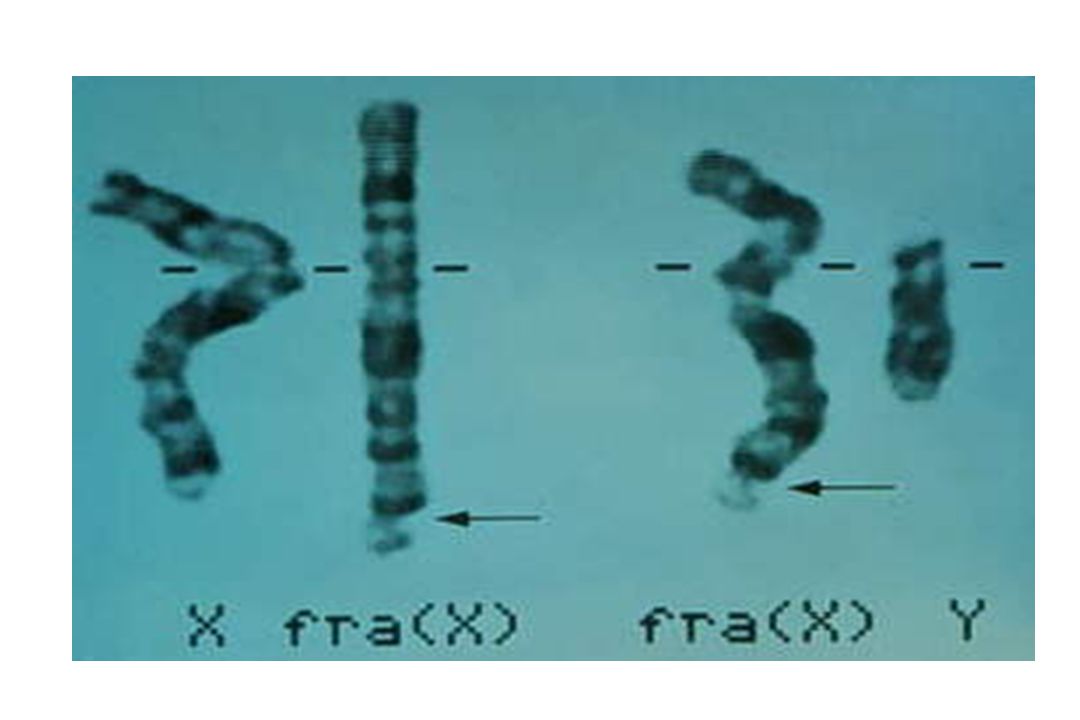
Exemple : syndrome du X fragile
Amplification de triplets CGG dans la région 5’UTR du gène FMR1 sujet N à 50 CGG sujet prémuté à 200 CGG sujet muté > 200 CGG + hyperméthylation entraînant une diminution d’expression du produit du gène et une perte de fonction 50 à 59 zone grise Le gène FMR1 est porté par le chromosome X Les garçons sont principalement touchés

38
EcoRI EcoRI EcoRI EcoRI EagI FMR1 (CGG)50 sonde EagI FMR1 (CGG)500
50 sonde EagI FMR1 (CGG)500")
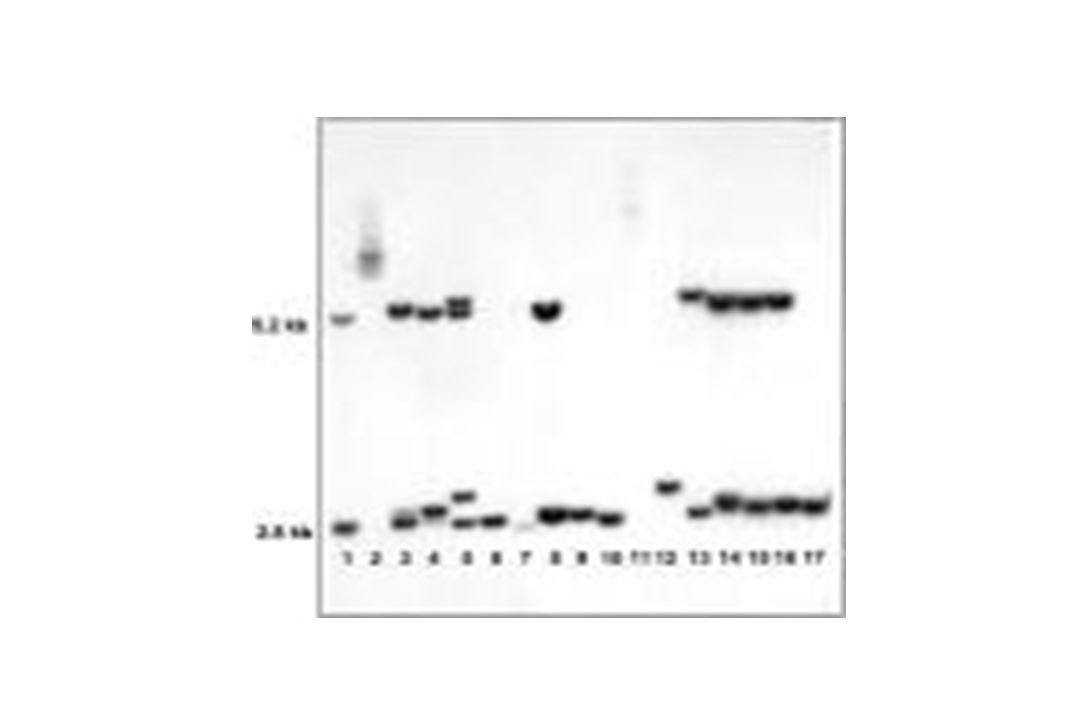
39
Coupure EcoRI + EagI N N PM PM MT MT 5,2 kb 2,8 kb MUTE PREMUTE
X INACTIF 5,2 kb NORMAL X INACTIF PREMUTE X ACTIF 2,8 kb NORMAL X ACTIF

42
B – Maladies à expansion de triplets « codants »
8 maladies neurodégénératives, toutes caractérisées par une expansion modérée d’une répétition CAG codant pour une Glutamine Signes cliniques apparaissent vers la quarantaine et évoluent vers un tableau de neurodégénérescence sévère - ces maladies sont dues à un mécanisme de « gain de fonction » lié à l’expression anormale de la région polyglutamine des protéines en cause Exemple : Chorée de Huntington

43
IV – Amplifications ou expansions de triplets
Maladie Nature du triplet Nombre de normal triplets pathologique Sd X fragile (CGG)n n=6-54 N > 200 Sd de Kennedy (CAG)n N=14-32 N > 40 Dystrophie myotonique de Steinert (CTG)n N=5-37 N > 50 Chorée de Huntington N=6-35 N > 37 SCA1 N=6-39 Retard mental FRAXE N=4-39 Sd de Jacobson N=11 N > 100 DRPLA N=3-36 N > 49 SCA3 N=13-44 N > 60 Ataxie de Friedreich (GAA)n N=6-29 SCA7 N=7-35 SCA2 N=13-32 N > 33 SCA6 N=4-18 N > 21
n. n=6-54. N > 200. Sd de Kennedy. (CAG)n. N= N > 40. Dystrophie myotonique. de Steinert. (CTG)n. N=5-37. N > 50. Chorée de Huntington. N=6-35. N > 37. SCA1. N=6-39. Retard mental FRAXE. N=4-39. Sd de Jacobson. N=11. N > 100. DRPLA. N=3-36. N > 49. SCA3. N= N > 60. Ataxie de Friedreich. (GAA)n. N=6-29. SCA7. N=7-35. SCA2. N= N > 33. SCA6. N=4-18. N > 21.")
44
CHOREE de HUNTINGTON - Maladie neurodégénérative de transmission autosomique dominante - Fréquence 1 / 20000 Mouvements involontaires associés à des troubles cognitifs et psy chiatriques. Perte des neurones cérébraux (IRM) essentiellement du striatum ( putamen ou noyau caudé) et de certaines couches cortex Début : 40 – 50 ans (pénétrance variable en fonction de l’âge) Répétition de (CAG) dans l’exon 1 de l’Huntingtine N =30 à 35 CAG Quand N > 35 chorée de Huntington Souvent hérité du père
 essentiellement. du striatum ( putamen ou noyau caudé) et de certaines couches cortex. Début : 40 – 50 ans (pénétrance variable en fonction de l’âge) Répétition de (CAG) dans l’exon 1 de l’Huntingtine. N =30 à 35 CAG. Quand N > 35 chorée de Huntington. Souvent hérité du père.")
45
IV – EPIMUTATIONS (Pathologie de l’épigénétique)
Anomalies de méthylation Anomalies de remodelage chromatinien Exemple : Syndrome ICF (immunodeficiency, centromeric instability and facial anomalies) : Régions d’hétérochromatine (chr.1/16) hypométhylées à cause d’une mutation d’une DNA méthylase Anomalies de l’empreinte génomique Exemple : Syndrome de Beckwith-Wiedemann PraderWilli Angelman
 : Régions d’hétérochromatine (chr.1/16) hypométhylées. à cause d’une mutation d’une DNA méthylase. Anomalies de l’empreinte génomique. Exemple : Syndrome de Beckwith-Wiedemann. PraderWilli. Angelman.")
Présentations similaires









>")
 Pr E. Tournier-Lasserve>")



 Pr E. Tournier-Lasserve>")



>")


