Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Epilepsies myocloniques progressives
D. Taussig Explorations Fonctionnelles CHU Rennes Septembre 05

2
Syndrome épileptique rare (1% des patients dans un centre spécialisé)
Epilepsies généralisées symptomatiques Maladies neurologiques peu fréquentes, avec souvent cause génétique. Au début, diagnostic différentiel avec épilepsies idiopathiques Au cours de l'évolution, problème du diagnostic étiologique Identification des causes : progrès incessants de la génétique moléculaire

3
Définitions Epilepsie myoclonique progressive : Lundborg (1903) (Unverricht,1891 : myoclonie). Depuis, syndrome de Ramsay Hunt : épilepsies myocloniques progressives (EMP) et ataxies myocloniques progressives (AMP). En commun : myoclonies, ataxie et détérioration intellectuelle progressives. En revanche, pas d’épilepsie dans les AMP Causes principales des AMP : dégénérescences spinocérébelleuses, maladies mitochondriales, certaines EMP au début (maladie d’Unverricht-Lundborg, sialidoses par exemple). Il semble plus clair de distinguer les deux syndromes.
 et ataxies myocloniques progressives (AMP). En commun : myoclonies, ataxie et détérioration intellectuelle progressives. En revanche, pas d’épilepsie dans les AMP. Causes principales des AMP : dégénérescences spinocérébelleuses, maladies mitochondriales, certaines EMP au début (maladie d’Unverricht-Lundborg, sialidoses par exemple). Il semble plus clair de distinguer les deux syndromes.")
4
Le syndrome d'épilepsie myoclonique progressive
myoclonies, crises d'épilepsie généralisées tonico-cloniques, ataxie et démence progressives, d'intensités variables selon les causes. Myoclonies toujours parcellaires et multifocales, souvent exacerbées par la posture, l'action ou des stimulus extérieurs comme la lumière, le son, le toucher. Le plus souvent, face et partie distale des membres, mais aussi muscles proximaux. Myoclonies le plus souvent corticales.

5
maladies où l’EMP n’est qu’un des éléments du tableau clinique
Age de début Maladie Petite enfance syndrome de Tay-Sachs, hyperglycémie non cétosique phénylcétonurie céroide lipofuscinose infantile précoce (CLN1) gangliosidoses à GM2 Enfance- Adolescence panencéphalite sclérosante subaigue maladie de Huntington juvénile maladie de Wilson Age adulte Atrophie dentato-rubro-pallido-luysienne maladie de Creutzfeldt-Jakob maladie d’Alzheimer, démence du syndrome de Down Syndrome myoclonies d’action-insuffisance rénale épilepsie myoclonique bénigne familiale de l’adulte
 gangliosidoses à GM2. Enfance- Adolescence. panencéphalite sclérosante subaigue. maladie de Huntington juvénile. maladie de Wilson. Age adulte. Atrophie dentato-rubro-pallido-luysienne. maladie de Creutzfeldt-Jakob. maladie d’Alzheimer, démence du syndrome de Down. Syndrome myoclonies d’action-insuffisance rénale. épilepsie myoclonique bénigne familiale de l’adulte.")
6
Caractéristiques cliniques des principales maladies responsables d'EMP
Transmis sion généti que Début possible à l'âge adulte Syndrome démentiel Signes associés Evolutivité Maladie d'Unverricht-Lundborg AR non mineur faible maladie de Lafora oui important crises d'épilepsie occipitales sévère syndrome MERFF sporadique ou maternelle variable surdité, myopathie, neuropathie, atrophie optique, accidents vasculaires céroides-lipofuscino ses AR ou AD (maladie de Kufs ) atrophie optique, dégénérescence maculaire sialidoses taches rouges cerises au fond d'oeil, opacités du cristallin
 atrophie optique, dégénérescence maculaire. sialidoses. taches rouges cerises au fond d oeil, opacités du cristallin.")
7
Maladie d'Unverricht-Lundborg
Début entre 6 et 15 ans Myoclonies sensibles aux stimuli au premier plan Progression lente, ataxie modérée, petite détérioration intellectuelle EEG : ralentissement activité de fond dès le début, pointes-ondes à 3-5 Hz avec amplitude maximale dans les régions centrales. Photosensibilité importante.

8
Maladie d'Unverricht-Lundborg
Début entre 6 et 15 ans Myoclonies sensibles aux stimuli au premier plan Progression lente, ataxie modérée, petite détérioration intellectuelle EEG : ralentissement activité de fond dès le début, pointes-ondes à 3-5 Hz avec amplitude maximale dans les régions centrales. Photosensibilité importante.

9
Maladie d'Unverricht-Lundborg (biologie moléculaire)
Autosomique récessive Plusieurs mutations dans le gène EPM1 ou CSTB Gène code pour un inhibiteur des cystéines-protéases, la cystatine B qui aurait un rôle neuroprotecteur

10
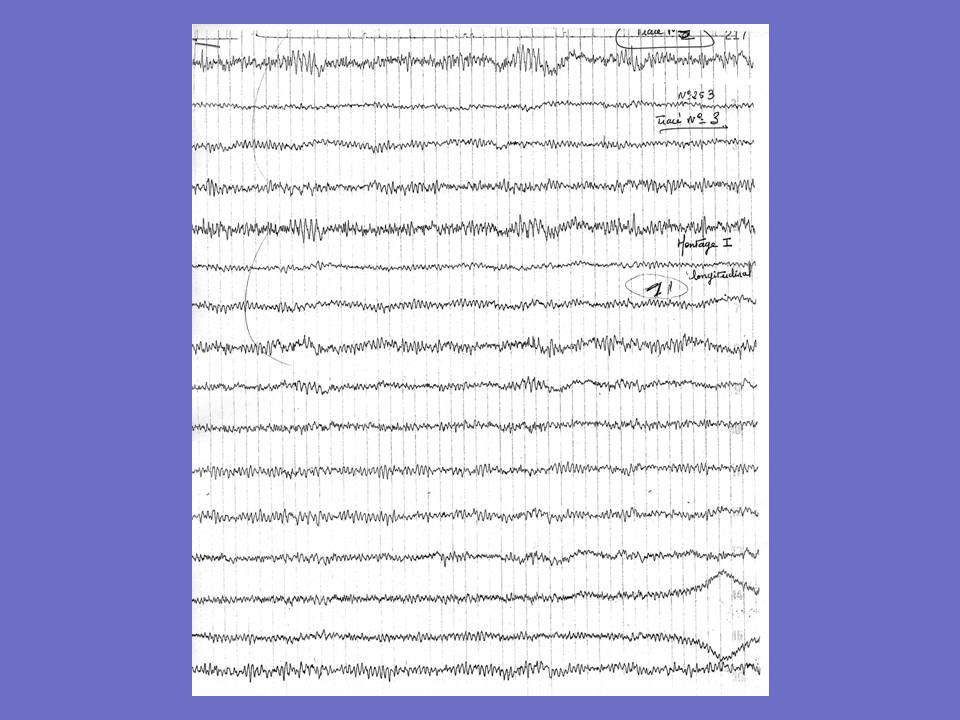
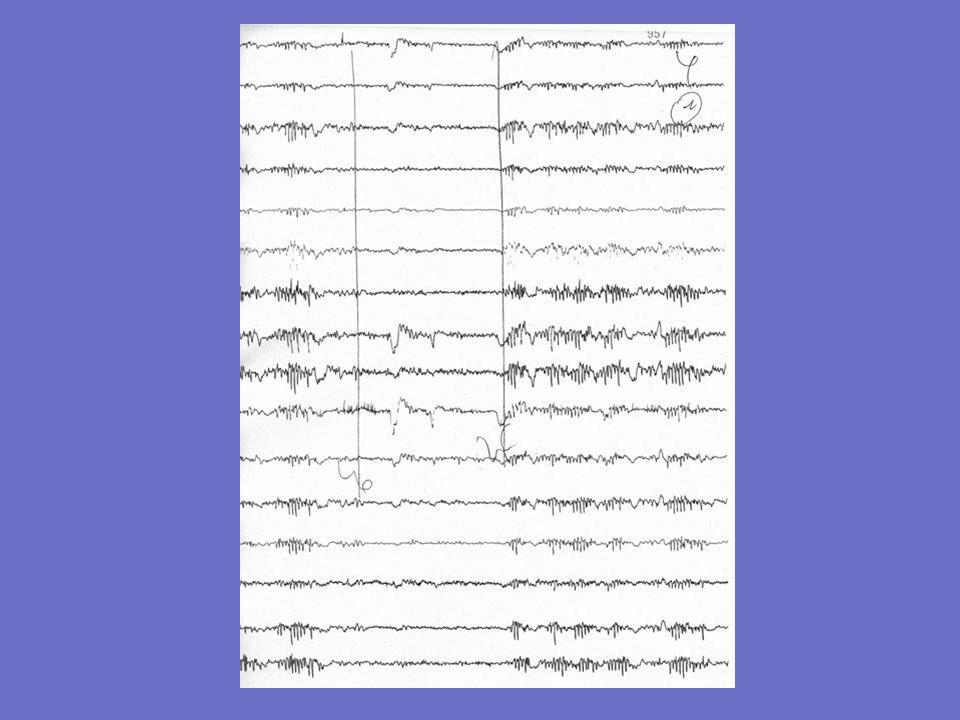
Exemples d ’EEG chez un patient né en 1958
1er tracé : 1989: 7 ans après le début 2è tracé: 1997: 15 ans après le début L ’EEG fait en 2000 est noté comme similaire Les tracés sont mal spatialisés; ondes lentes bifrontales sur le 1er, RAS par ailleurs

13
Maladie de Lafora Début entre 9 et 18 ans
Mode de début : myoclonies; crise généralisée tonico-clonique ; difficultés scolaires ; syndrome dépressif, hallucinations visuelles (crises occipitales) Tableau se complète : myoclonies permanentes, ataxie cérébelleuse, démence. Décès dans les 10 ans EEG : Au début : rythme de fond ralenti, poly PO irrégulières souvent augmentées par la stimulation lumineuse intermittente (SLI) mais pas pendant le sommeil. Puis détérioration du rythme de fond et disparition des rythmes de sommeil, sommeil paradoxal seul identifié. Ensuite anomalies épileptiques multifocales à prédominance postérieure A la phase terminale, désorganisation complète de l’EEG
 Tableau se complète : myoclonies permanentes, ataxie cérébelleuse, démence. Décès dans les 10 ans. EEG : Au début : rythme de fond ralenti, poly PO irrégulières souvent augmentées par la stimulation lumineuse intermittente (SLI) mais pas pendant le sommeil. Puis détérioration du rythme de fond et disparition des rythmes de sommeil, sommeil paradoxal seul identifié. Ensuite anomalies épileptiques multifocales à prédominance postérieure. A la phase terminale, désorganisation complète de l’EEG.")
14
Maladie de Lafora Corps de Lafora dans cerveau, foie, muscle
Dans les neurones : péricaryons et dendrites Colorés par PAS; accumulation de polyglucosans (polysaccharides) Diagnostic: biopsie cutanée dans le creux axillaire
 Diagnostic: biopsie cutanée dans le creux axillaire.")
15
Maladie de Lafora (biologie moléculaire 1)
Autosomique récessive Mutations dans gène EPM2A : code pour laforine (48% des cas), tyrosine phosphatase associée aux ribosomes; rôle dans la traduction. Peut aussi provoquer un hyperfonctionnement de glycogène synthase (corps de Lafora) Identification de protéines partenaires de laforine
, tyrosine phosphatase associée aux ribosomes; rôle dans la traduction. Peut aussi provoquer un hyperfonctionnement de glycogène synthase (corps de Lafora) Identification de protéines partenaires de laforine.")
16
Maladie de Lafora (biologie moléculaire 2)
Mutations dans gène EPM2B ou NHLRC1 : code pour maline (40% des cas), impliquée dans la protéolyse Au moins un autre gène (12% des cas)
, impliquée dans la protéolyse. Au moins un autre gène (12% des cas)")
17
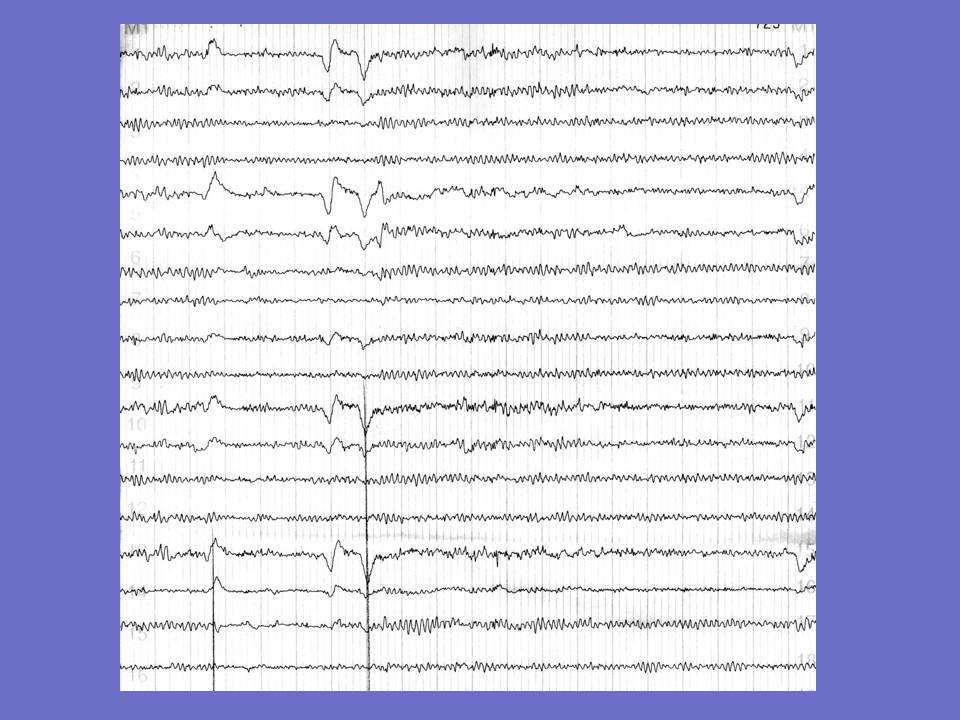
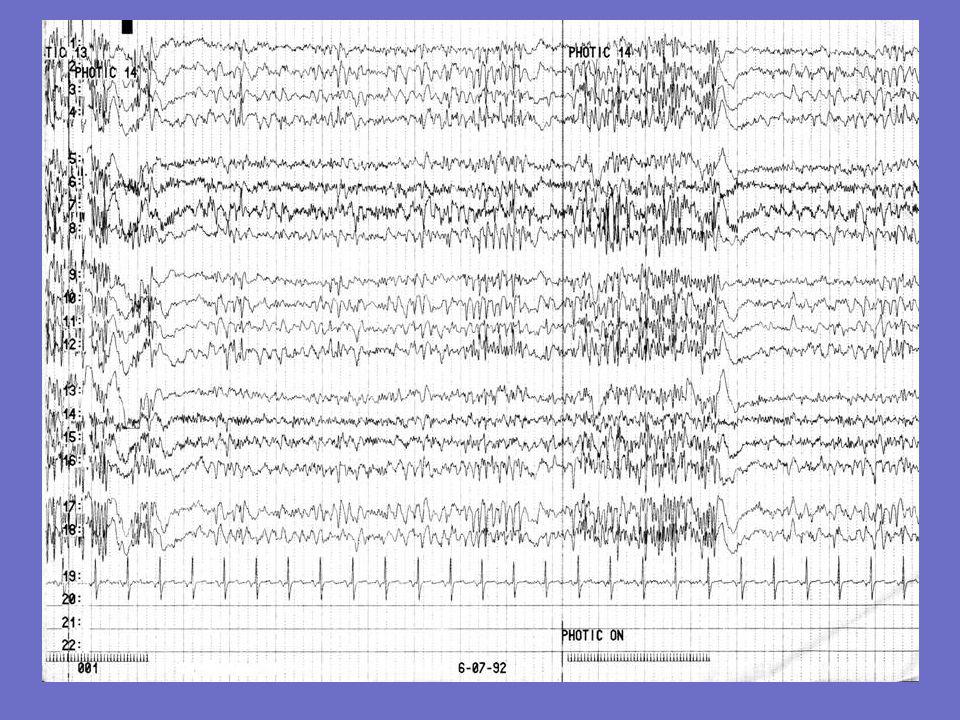
Exemples d ’EEG chez un patient né en 1982
1er tracé: 1997: après une 1ère crise. Mal spatialisé, ne réagissant pas à l ’ouverture des yeux. Bouffées de pointes-ondes et polypointes-ondes généralisées. 2è tracé: 2001: beaucoup de myoclonies Pas de rythmes de fond; fréquentes bouffées de polypointes-ondes généralisées 3è tracé : janvier 2003: myoclonies permanentes, grabataire. Etat de mal électrique Décès en août 2004

21
Maladies mitochondriales (MERRF)
Présentation clinique, âge de début et sévérité de l'atteinte extrêmement variables Fréquemment surdité, myopathie,neuropathie, atrophie optique, rétinopathie. Certains à la frontière avec le syndrome MELAS EEG : ralentissement global du rythme de fond, décharges de PO généralisées entre 2 et 5 Hz, photosensibilité. Dosages des enzymes de la chaîne respiratoire et biopsie musculaire peuvent être normaux Mutation la plus fréquente : mutation ponctuelle de paire de base 8344 du gène de l’ARNt mitochondrial de la lysine. Plus rarement, paire de base 8356 du gène du même ARNt mutée. Dans d'autres familles, mutations ponctuelles (plusieurs types) dans l’ARNt mitochondrial de la sérine
 dans l’ARNt mitochondrial de la sérine.")
22
Céroides-lipofuscinoses
Dépôt de lipopigments autofluorescents lysosomiaux dans le système nerveux central et d’autres organes. Sous-unité C de l'ATP synthase mitochondriale est le composant principal du dépôt Six types génétiquement distincts (chez le nourrisson, l’enfant ou l’adulte), groupés dans le monde en foyers tous de transmission autosomique récessive sauf la maladie de Kufs qui peut être autosomique dominante. Les formes de l'enfant sont souvent regroupées sous le terme de syndrome de Batten. Diagnostic de certitude : étude en microscopie électronique de la biopsie de peau. Inclusions dans les cellules sécrétrices eccrines caractéristiques
, groupés dans le monde en foyers tous de transmission autosomique récessive sauf la maladie de Kufs qui peut être autosomique dominante. Les formes de l enfant sont souvent regroupées sous le terme de syndrome de Batten. Diagnostic de certitude : étude en microscopie électronique de la biopsie de peau. Inclusions dans les cellules sécrétrices eccrines caractéristiques.")
23
Céroides-lipofuscinoses : type infantile tardif (1)
Maladie de Jansky-Bielschowsky : NCL type 2 Début entre 1 et 4 ans Troubles de la marche avec ataxie, troubles du langage et régression psychomotrice, puis crises d’épilepsie avec myoclonies. Cécité progressive avec atrophie optique avant l’âge de 5 ans. Décès entre 3 et 10 ans.

24
Céroides-lipofuscinoses : type infantile tardif (2)
EEG : ralentissement de l’activité de fond, surcharge lente irrégulière et décharges de polypointes ondes. SLI à basse fréquence induit des pointes polyphasiques postérieures très amples. Gène TPP1 situé sur le chromosome 11p15 code pour la tripeptidyl peptidase 1 (enlève les tripeptides du N-terminal des protéines dégradées dans les lysosomes). Différentes mutations Variants NCL type 5 (gène CLN5) et 6 (gène CLN6)
. Différentes mutations. Variants NCL type 5 (gène CLN5) et 6 (gène CLN6)")
25
Céroides-lipofuscinoses : forme juvénile de Spielmeyer-Vogt-Sjögren (type 3) (1)
répandue en Scandinavie. début entre 4 et 15 ans : baisse d’acuité visuelle liée à dégénérescence maculaire. Atteinte intellectuelle lente et progressive. Début des signes neurologiques après 2 à 3 ans avec syndrome extrapyramidal puis ataxie et atteinte pyramidale. Absences et surtout crises tonico-cloniques et myoclonies segmentaires (en particulier des muscles faciaux) 1 à 4 ans après le début. Plus tardivement myoclonies massives exacerbées par la mobilisation passive. Episodes psychotiques. Décès vers l’âge de 20 ans, souvent dans un état de mal myoclonique.
 1 à 4 ans après le début. Plus tardivement myoclonies massives exacerbées par la mobilisation passive. Episodes psychotiques. Décès vers l’âge de 20 ans, souvent dans un état de mal myoclonique.")
26
Céroides-lipofuscinoses : forme juvénile de Spielmeyer-Vogt-Sjögren (type 3) (2)
EEG : ralentissement précoce du rythme de fond et décharges paroxystiques parfois de longue durée, sans photosensibilité. Aspect vacuolisé des lymphocytes Gène situé sur le chromosome 16p12 : CLN3 : code pour une grosse protéine hydrophobe; différentes mutations décrites.

27
Céroides-lipofuscinoses : maladie de Kufs (CLN4)
débute entre 11 et 50 ans et évolue vers le décès en 10 ans. Certaines formes se caractérisent par une démence plutôt que par un syndrome complet d'EMP. EEG altéré uniquement dans les formes avec EMP avec un rythme de fond ralenti, des pointes-ondes généralisées et une réponse intense à la SLI à basse fréquence. biopsie cérébrale pour mettre en évidence les inclusions (dans la peau dépôts de lipofuschine physiologiques liés à l’âge).
.")
28
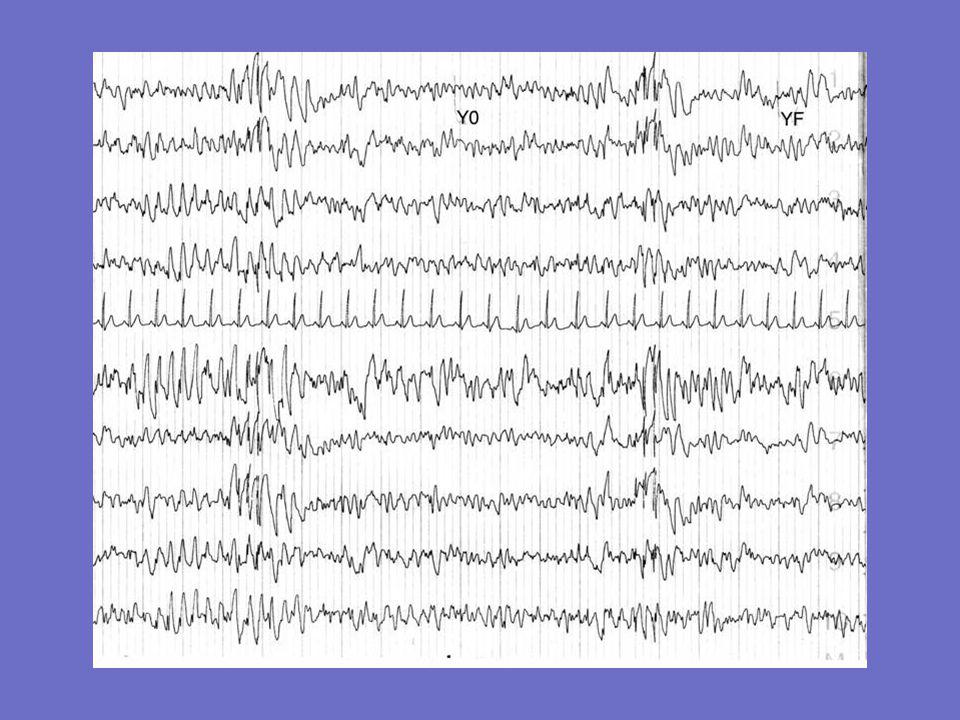
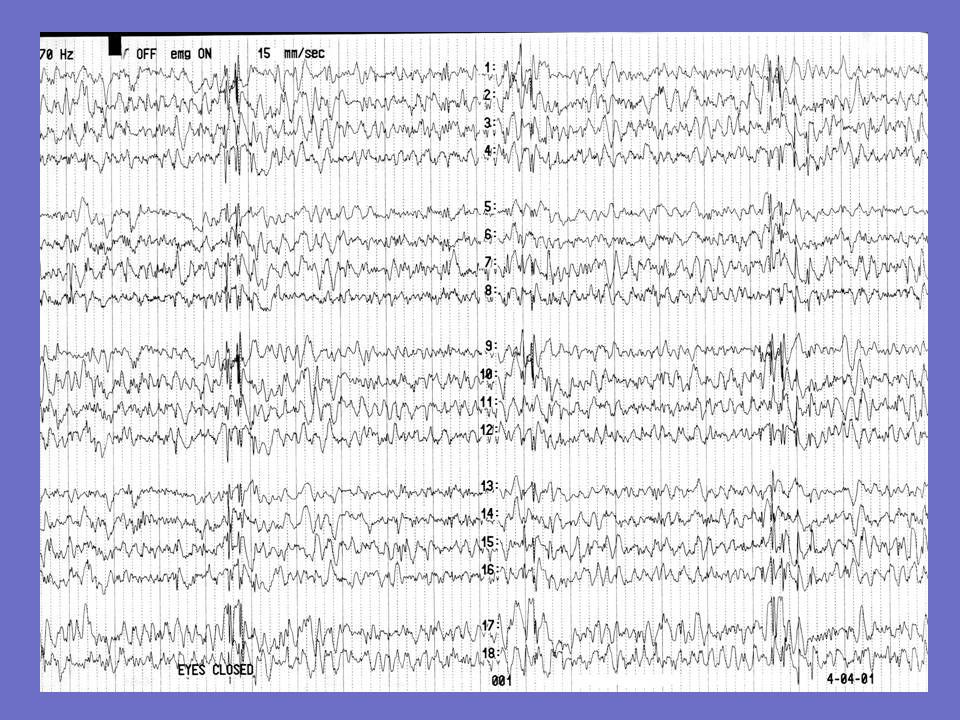
Exemple d ’EEG chez une patiente née en 1943
Début en 1968 1er tracé 1991: bouffées de polypointes à prédominance postérieure disparaissant à l ’ouverture des yeux 2è tracé: 1992: photosensibilité décès en 1994 (tuberculose)
")
31
Sialidoses (1) maladies autosomiques récessives associées à un déficit en N-acetylneuraminidase lysosomiale type 1 : Italie; gène NEU1 sur le chromosome 6p21-3, différentes mutations Le plus souvent, début entre 8 et 15 ans, parfois à l’âge adulte. Déficit visuel modéré avec anomalies ophtalmologiques caractéristiques : taches rouges cerises au fond d’oeil et opacités cristalliniennes punctiformes. Douleurs des extrémités, myoclonies faciales spontanées, irrégulières, prédominant au niveau de la bouche, peu sensibles aux stimulations, persistent pendant le sommeil.

32
Sialidoses (2) EEG : activité de fond normale ou rapide sans photosensibilité. Les myoclonies massives sont associées à des décharges de pointes-ondes généralisées. Diagnostic de certitude : déficit en neuraminidase dans les lymphocytes et fibroblastes en culture et augmentation de l'élimination urinaire de sialoliposaccharides. type 2: Japon : galactosialidoses (déficit en une protéine indispensable à l’action de la neuraminidase); dysmorphie
; dysmorphie.")
33
Maladie de Gaucher de type 3
Début dans l’enfance, à l’adolescence ou à l’âge adulte EMP sans démence, mais avec des troubles des saccades oculaires, une paralysie supranucléaire Splénomégalie Evolution très sévère ou moins Pancytopénie et augmentation des phosphatases acides sériques. Autosomique récessive Activité -glucocérébrosidase basse dans les lymphocytes ou les fibroblastes en culture. Stockage des glucocérébrosides au niveau des lymphocytes circulants et des cellules de la moelle osseuse. Diagnostic prénatal possible sur les cellules amniotiques. Nombreuses mutations décrites.

34
Répartition: l ’expérience du centre Saint-Paul (1960-2004)
")
35
Diagnostic de certitude des principales maladies responsables d'EMP
Examen paraclinique Génétique moléculaire maladie d'Unverricht-Lundborg Aucun mutations du gène de la cystatine (EPM1 ou CSTB) maladie de Lafora biopsie cutanée dans le creux axillaire (corps de Lafora) mutations des gènes EPM2A (laforine) EPM2B ou NHLRC1 (maline) autre syndrome MERFF biopsie musculaire, dosage enzymes de la chaîne respiratoire mutations de l’ARNt mitochondrial de la lysine (MTTK) et de la sérine céroïdes-lipofuscinoses biopsie cutanée : étude en microscopie électronique des inclusions biopsie cérébrale (maladie de Kufs) mutations des gènes de TPP1, CLN3, CLN5, CLN6 sialidoses type 1: déficit en neuraminidase dans les lymphocytes et fibroblastes en culture augmentation de l'élimination urinaire de sialoliposaccharides. - mutations du gène NEU1 (type 1) locus sur le chromosome 20 (type 2)
 maladie de Lafora. biopsie cutanée dans le creux axillaire (corps de Lafora) mutations des gènes. EPM2A (laforine) EPM2B ou NHLRC1 (maline) autre. syndrome MERFF. biopsie musculaire, dosage enzymes de la chaîne respiratoire. mutations de l’ARNt mitochondrial de la lysine (MTTK) et de la sérine. céroïdes-lipofuscinoses. biopsie cutanée : étude en microscopie électronique des inclusions. biopsie cérébrale (maladie de Kufs) mutations des gènes de TPP1, CLN3, CLN5, CLN6. sialidoses. type 1: déficit en neuraminidase dans les lymphocytes et fibroblastes en culture augmentation de l élimination urinaire de sialoliposaccharides. - mutations du gène NEU1 (type 1) locus sur le chromosome 20 (type 2)")
36
Traitement symptomatique des EMP: les antiépileptiques
Médicaments classiques : valproate (sauf pour le MERRF), benzodiazépines Médicaments nouveaux : topiramate, levetiracetam, zonisamide (ATU) A proscrire : carbamazépine, phénytoine ??? : lamotrigine piracetam (myoclonies)
, benzodiazépines. Médicaments nouveaux : topiramate, levetiracetam, zonisamide (ATU) A proscrire : carbamazépine, phénytoine. : lamotrigine. piracetam (myoclonies)")
37
Traitement symptomatique des EMP
kinésithérapie ; prises en charge psychologique et sociale Associations Conseil génétique

38
Traitements étiologiques
N acétylcystéine dans maladie d’Unverricht-Lundborg: antioxydant : per os au long cours. Effets variables selon études sur épilepsie et ataxie Régime cétogène dans la maladie de Lafora ; étude en cours L-carnitine et Coenzyme Q dans les maladies mitochondriales Enzyme recombinante dans maladie de Gaucher : pas d’effet sur les signes neurologiques

39
Associations de patients
Association contre la maladie rare myoclonique d'Unverricht-Lundborg Président : M. José MARTY Adresse : Maison-Neuve SAINT-CHAMASSY Téléphone +33 (0) Association France Lafora Président : M. Etienne HAMEAU Adresse :16 Rue du docteur Jules Amaudrut LAVAL Téléphone +33 (0)
 Association France Lafora Président : M. Etienne HAMEAU Adresse :16 Rue du docteur Jules Amaudrut LAVAL Téléphone +33 (0)")
40
Quelques références Aykutlu et al. (2005) Add-on therapy with topiramate in progressive myoclonic epilepsy. Epilepsy and behavior, 6, Badhwar et al. (2004) Action myoclonus-renal failure syndrome: characterizatrion of a unique cerebro-renal disorder. Brain, 127, Chan E, Andrade D, Franceschetti S, Minassian B (2005) Progressive myoclonus epilepsies: EMP1, EMP2A, EMP2B. Adv Neurol, 95, Crest et al. (2004) Levetiracetam in progressive myoclonic epilepsy. Neurology, 62, Edwards et al. (2002) N-acetylcysteine and Unverricht-Lundborg disease: variable response and possible side effects. Neurology, 59, Falco et al. (2003) Benign adult familial myoclonic epilepsy. Genetic heterogeneity and alleleism with ADCME. Neurology, 60, ? Genton P, Bureau M (2004). Les épilepsies myovcloniques progressives: mythe ou réalité? Epileptic Disord ; 6: S139-46 Goebel HH, Sharp JD (1998) The neuronal ceroid-lipofuscinoses. Recent advences. Brain Pathol, 8, Gomez-Abad et al. (2005) Lafora disease due to EPM2B mutations. A clinical and genetic study. Neurology, 64, Kyllerman M, Ben-Menachem E (1998) Zonisamide for progressive myoclonus epilepsy : long-term observation in seven patients. Epilepsy Res, 29, Leppick et al.(2003) Classification of the myoclonic epilepsies. Epilepsia, 44, 2-6. Magaudda et al. (2004) Antimyoclonic effect of levetiracetam in 13 patients with Unverrricht-Lundborg disease: clinical observations. Epilepsia, 45, Park et al. (2003) Myoclonic epilepsy in Gaucher disease : genotype-phenotype insights from a rare patient subgroup. Pediatric research, 53, Roger J, Genton P, et al. (1992) Les épilepsies myocloniques progressives de l'enfant et l'adolescent. In: Les syndromes épileptiques de l’enfant et l’adolescent. Edited by M. Bureau, C. Dravet, F. E. Dreifuss, A. Perret and P. Wolf. Londres: John Libbey & Company Ltd, pp Shahwan et al. (2005) Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol, 4,
 Action myoclonus-renal failure syndrome: characterizatrion of a unique cerebro-renal disorder. Brain, 127, Chan E, Andrade D, Franceschetti S, Minassian B (2005) Progressive myoclonus epilepsies: EMP1, EMP2A, EMP2B. Adv Neurol, 95, Crest et al. (2004) Levetiracetam in progressive myoclonic epilepsy. Neurology, 62, Edwards et al. (2002) N-acetylcysteine and Unverricht-Lundborg disease: variable response and possible side effects. Neurology, 59, Falco et al. (2003) Benign adult familial myoclonic epilepsy. Genetic heterogeneity and alleleism with ADCME. Neurology, 60, Genton P, Bureau M (2004). Les épilepsies myovcloniques progressives: mythe ou réalité Epileptic Disord ; 6: S Goebel HH, Sharp JD (1998) The neuronal ceroid-lipofuscinoses. Recent advences. Brain Pathol, 8, Gomez-Abad et al. (2005) Lafora disease due to EPM2B mutations. A clinical and genetic study. Neurology, 64, Kyllerman M, Ben-Menachem E (1998) Zonisamide for progressive myoclonus epilepsy : long-term observation in seven patients. Epilepsy Res, 29, Leppick et al.(2003) Classification of the myoclonic epilepsies. Epilepsia, 44, 2-6. Magaudda et al. (2004) Antimyoclonic effect of levetiracetam in 13 patients with Unverrricht-Lundborg disease: clinical observations. Epilepsia, 45, Park et al. (2003) Myoclonic epilepsy in Gaucher disease : genotype-phenotype insights from a rare patient subgroup. Pediatric research, 53, Roger J, Genton P, et al. (1992) Les épilepsies myocloniques progressives de l enfant et l adolescent. In: Les syndromes épileptiques de l’enfant et l’adolescent. Edited by M. Bureau, C. Dravet, F. E. Dreifuss, A. Perret and P. Wolf. Londres: John Libbey & Company Ltd, pp Shahwan et al. (2005) Progressive myoclonic epilepsies: a review of genetic and therapeutic aspects. Lancet Neurol, 4,")
Présentations similaires



>")



>")



 Pr E. Tournier-Lasserve>")

>")
>")





>")