Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
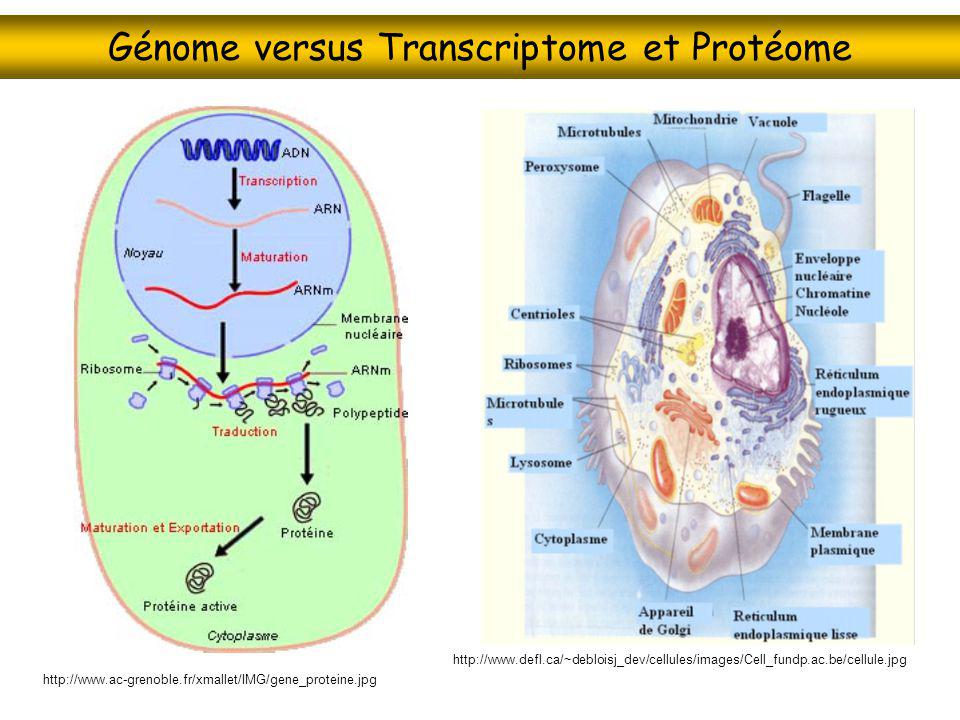
Protéomique Protéome = ensemble des protéines d’une cellule, ou d’une organelle, à un instant donné (et donc sous des conditions données) Connaissance du génome n’implique pas la connaissance du protéome (régulation de la transcription, de la traduction, de la localisation cellulaire, etc.) Connaissance du transcriptome n’implique pas la connaissance du protéome (régulation de la traduction et de la localisation cellulaire, durée de vie des protéines, etc.) Schizosaccharomyces pombe
 Connaissance du transcriptome n’implique pas la connaissance du protéome (régulation de la traduction et de la localisation cellulaire, durée de vie des protéines, etc.) Schizosaccharomyces pombe.")
2
Protéomique Génome = ensemble des gènes d’un organisme, ou d’une espèce Protéome = ensemble des protéines d’une cellule, ou d’une organelle, à un instant donné (et donc sous des conditions données) Y-a-t-il un parallèle complet entre génomique et protéomique ? La protéomique permet de quantifier les variations de leur taux d'expression en fonction du temps, de leur environnement, de leur état de développement, de leur état physiologique et pathologique, de l'espèce d'origine. Elle étudie aussi les interactions que les protéines ont avec d'autres protéines, avec l'ADN ou l'ARN, avec des substances. La protéomique fonctionnelle étudie les fonctions de chaque protéine. La protéomique étudie enfin la structure primaire, secondaire et tertiaire des protéines
 Y-a-t-il un parallèle complet entre génomique et protéomique La protéomique permet de quantifier les variations de leur taux d expression en fonction du temps, de leur environnement, de leur état de développement, de leur état physiologique et pathologique, de l espèce d origine. Elle étudie aussi les interactions que les protéines ont avec d autres protéines, avec l ADN ou l ARN, avec des substances. La protéomique fonctionnelle étudie les fonctions de chaque protéine. La protéomique étudie enfin la structure primaire, secondaire et tertiaire des protéines.")
3
Génome versus Transcriptome et Protéome
4
Protéomique Génome = ensemble des gènes d’un organisme, ou d’une espèce Protéome = ensemble des protéines d’une cellule, ou d’une organelle, à un instant donné (et donc sous des conditions données) Y-a-t-il un parallèle complet entre génomique et protéomique ? Génome : taux d’erreur de la réplication, ~10-7 modifications de la chromatine (méthylation, etc.) Protéome : taux d’erreur de la transcription, erreur/polymorphisme de l’épissage erreur/polymorphisme de l’édition erreur de la traduction, 10-4 repliement modifications post-traductionnelles (irréversible/réversibles) localisation cellulaire La protéomique permet de quantifier les variations de leur taux d'expression en fonction du temps, de leur environnement, de leur état de développement, de leur état physiologique et pathologique, de l'espèce d'origine. Elle étudie aussi les interactions que les protéines ont avec d'autres protéines, avec l'ADN ou l'ARN, avec des substances. La protéomique fonctionnelle étudie les fonctions de chaque protéine. La protéomique étudie enfin la structure primaire, secondaire et tertiaire des protéines
 Y-a-t-il un parallèle complet entre génomique et protéomique Génome : taux d’erreur de la réplication, ~10-7. modifications de la chromatine (méthylation, etc.) Protéome : taux d’erreur de la transcription, erreur/polymorphisme de l’épissage. erreur/polymorphisme de l’édition. erreur de la traduction, repliement. modifications post-traductionnelles (irréversible/réversibles) localisation cellulaire. La protéomique permet de quantifier les variations de leur taux d expression en fonction du temps, de leur environnement, de leur état de développement, de leur état physiologique et pathologique, de l espèce d origine. Elle étudie aussi les interactions que les protéines ont avec d autres protéines, avec l ADN ou l ARN, avec des substances. La protéomique fonctionnelle étudie les fonctions de chaque protéine. La protéomique étudie enfin la structure primaire, secondaire et tertiaire des protéines.")
5
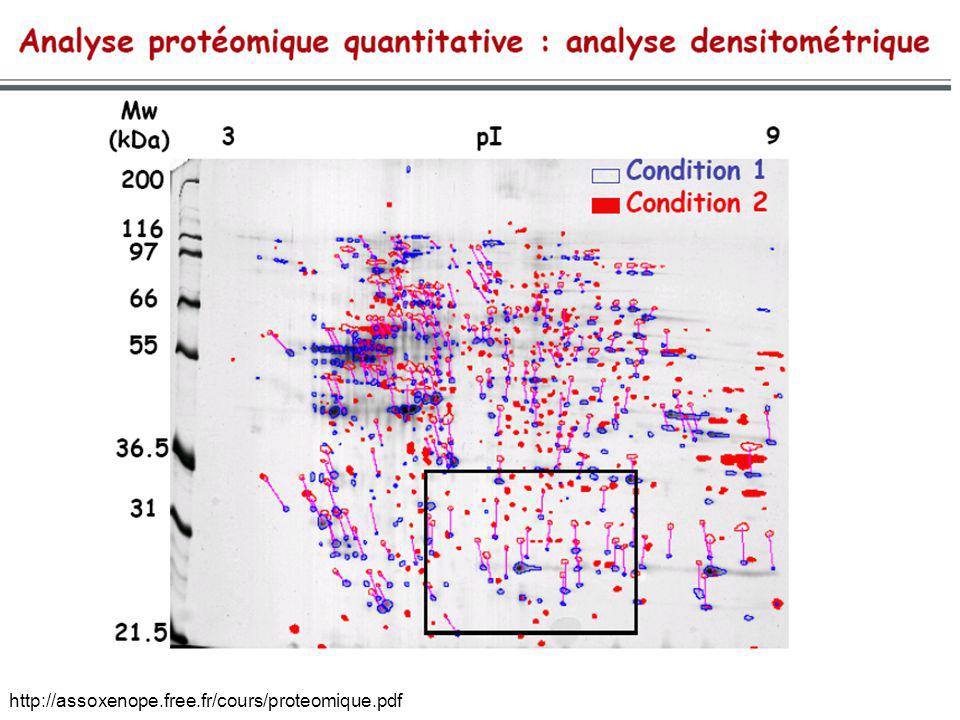
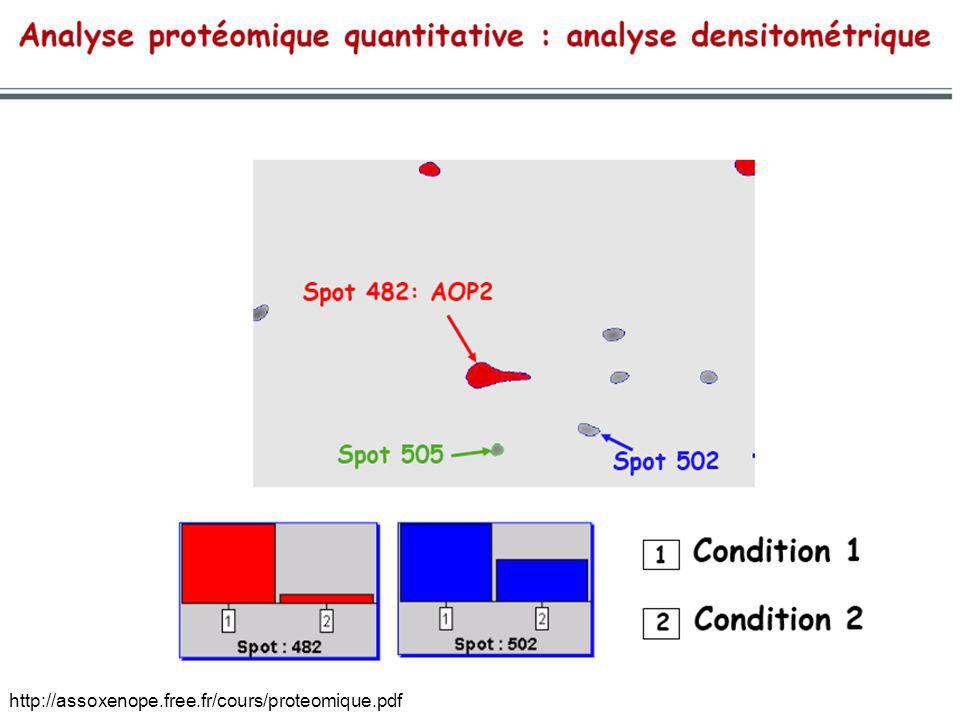
Électrophorèse 2D

6
Electrophorèse 2D

7
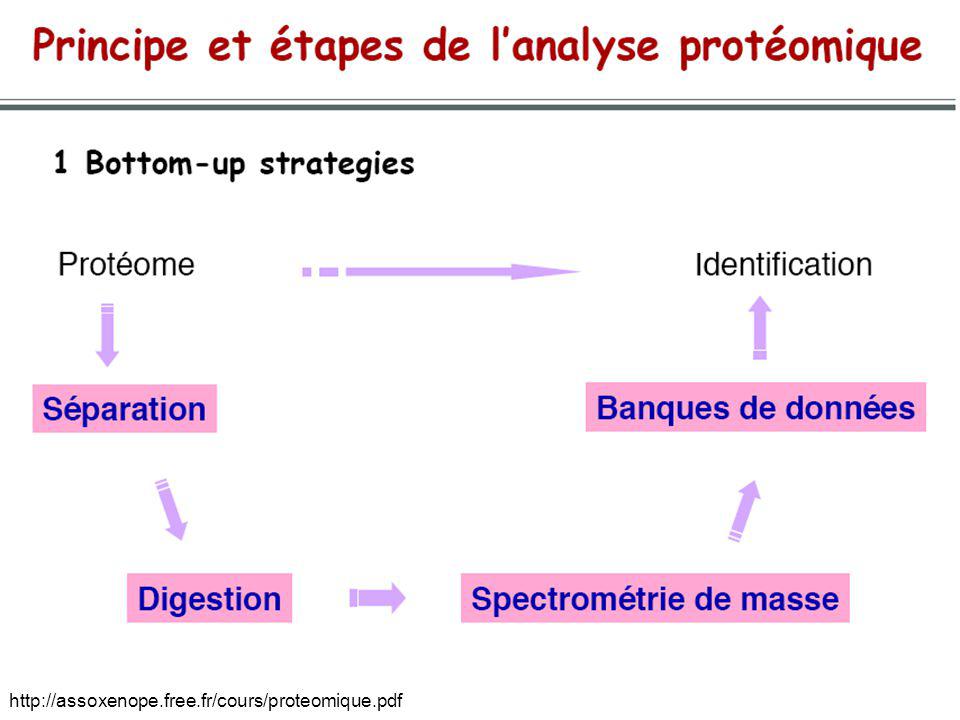
DIFFÉRENTES VISIONS DE LA PROTÉOMIQUE
Protéomique fonctionnelle : interactions protéines-protéines (double-hybride, PCA, Tap-Tag MS-MS, etc.), etc. Protéomique structurale : structure tridimensionnelle de toutes les protéines (cristallographie, RMN, etc.) Pharmacoprotéomique Etc. --> description de toutes les protéines à un instant t en utilisant la spectrométrie de masse
, etc. Protéomique structurale : structure tridimensionnelle de toutes les protéines (cristallographie, RMN, etc.) Pharmacoprotéomique. Etc. --> description de toutes les protéines à un instant t en utilisant la spectrométrie de masse.")
15
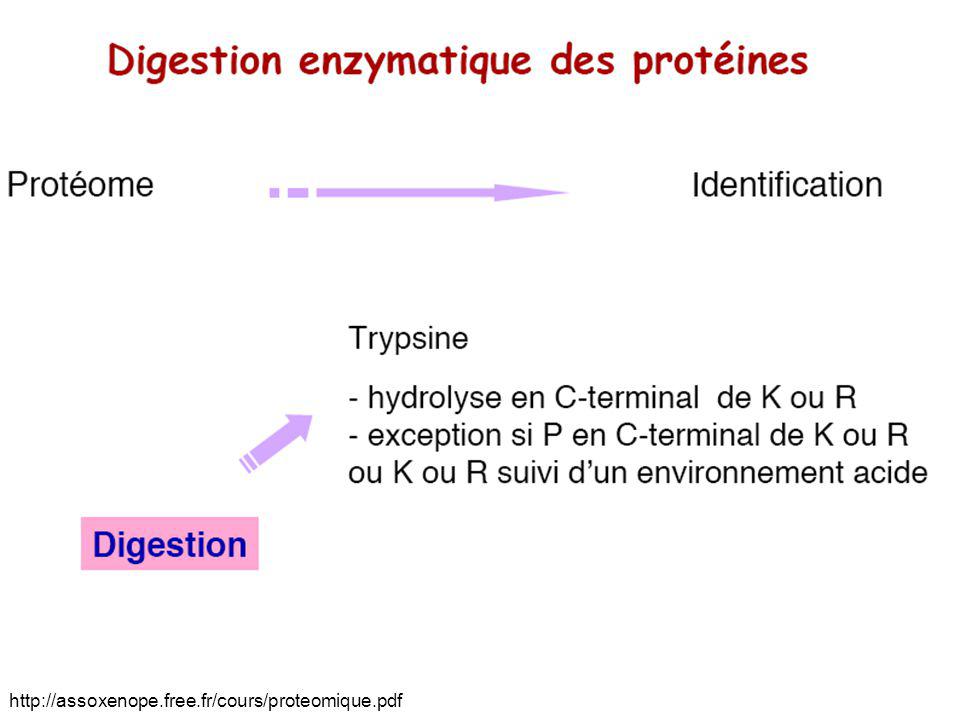
le NH2 terminal et le NH2 de la chaîne latérale de la Lys ou de l’Arg
Le peptide trypsique Se termine par une Lysine ou une Arginine donc 2 sites basiques protonables par peptide : le NH2 terminal et le NH2 de la chaîne latérale de la Lys ou de l’Arg R1 R3 O O C NH C C NH C NH3+ C C OH C NH C O R2 O CH2 (CH2)3 NH3+ Lysine coursenligne.u-strasbg.fr/depotcel/DepotCel/279/Intranet/Carapito.ppt
3. NH3+ Lysine. coursenligne.u-strasbg.fr/depotcel/DepotCel/279/Intranet/Carapito.ppt.")
19
Désorption-ionisation laser assistée par matrice
Matrix-Assisted Laser Desoption/Ionisation (MALDI)
")
20
Désorption-ionisation laser assistée par matrice (MALDI)
Avantages : * Possibilité d'ioniser des molécules de hautes masses moléculaires * Méthode d'ionisation douce : peu de fragmentation des ions moléculaires * Permet d'analyser des échantillons de faible concentration (de l'ordre de la picomole (10-12) et de la femtomole (10-15) ) * Produit principalement des ions monochargés, spectre plus simple à analyser * Grande tolérance aux sels et aux tampons Tampon et sels Masse molaire (en g/mol) Concentration maximale compatible avec le MALDI (en mM) Tris 121 100 HEPES 238 Bicine 163 50 Urée 60 500 Guanidine 96 250 DTT 154 Glycérol 92 130 PEG 2000 2000 0,5 Triton X-100 628 1,6 NP 40 603 1,7 SDS 288 0,35 Inconvénients : * Formation d'adduits (combinaison directe de 2 espèces chimiques distinctes) et d'ions de matrice
 et de la femtomole (10-15) ) * Produit principalement des ions monochargés, spectre plus simple à analyser. * Grande tolérance aux sels et aux tampons. Tampon et sels. Masse molaire (en g/mol) Concentration maximale compatible avec le MALDI (en mM) Tris HEPES Bicine Urée Guanidine DTT Glycérol PEG ,5. Triton X ,6. NP ,7. SDS ,35. Inconvénients : * Formation d adduits (combinaison directe de 2 espèces chimiques distinctes) et d ions de matrice.")
21
Ionisation par électrospray
Electrospray Ionisation (ESI) Pression élevée Pression faible Avantages : molécules en solution La multicharge permet l’étude de molécule de plus haut poids moléculaire que la limite de l'analyseur Inconvénients : complique l'analyse de spectre [M+nH]n+ n pouvant aller jusqu'à plusieurs dizaines multichargé : permet étude de molécule de plus haute poids moléculaire que limite de l'analyseur (par exemple si limite d'un analyseur est 2000 Da, il est possible de mesurer des molécules de Da avec un état de charge 20+). m/z = (M+3)/3, (M+4)/4, (M+5)/5, (M+6)/6
 Pression élevée. Pression faible. Avantages : molécules en solution. La multicharge permet l’étude de molécule de plus haut poids moléculaire que la limite de l analyseur. Inconvénients : complique l analyse de spectre. [M+nH]n+ n pouvant aller jusqu à plusieurs dizaines. multichargé : permet étude de molécule de plus haute poids moléculaire que limite de l analyseur (par exemple si limite d un analyseur est 2000 Da, il est possible de mesurer des molécules de Da avec un état de charge 20+). m/z = (M+3)/3, (M+4)/4, (M+5)/5, (M+6)/6.")
23
Temps de vol Time Of Flight (TOF) m étant la masse v la vitesse
l la distance parcourue pendant le vol t le temps de vol z la charge de l’ion V la tension accélératrice e étant la charge élémentaire

24
Le temps de vol L'analyseur quadripolaire Le piège ionique quadripolaire Le FT-ICR L'orbitrappe L'analyseur à secteur magnétique

25
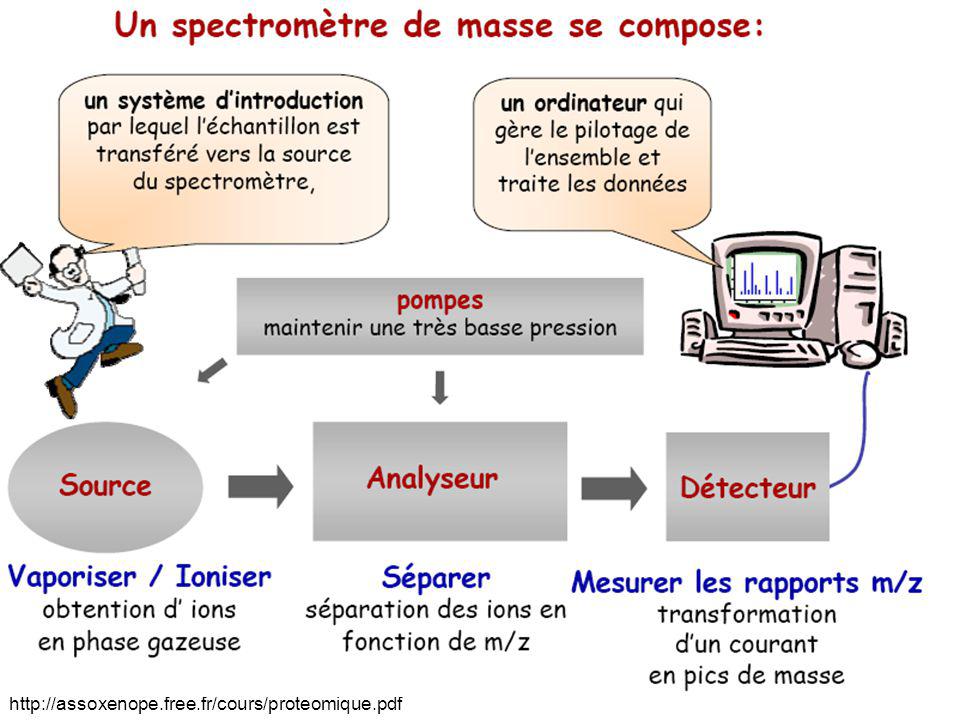
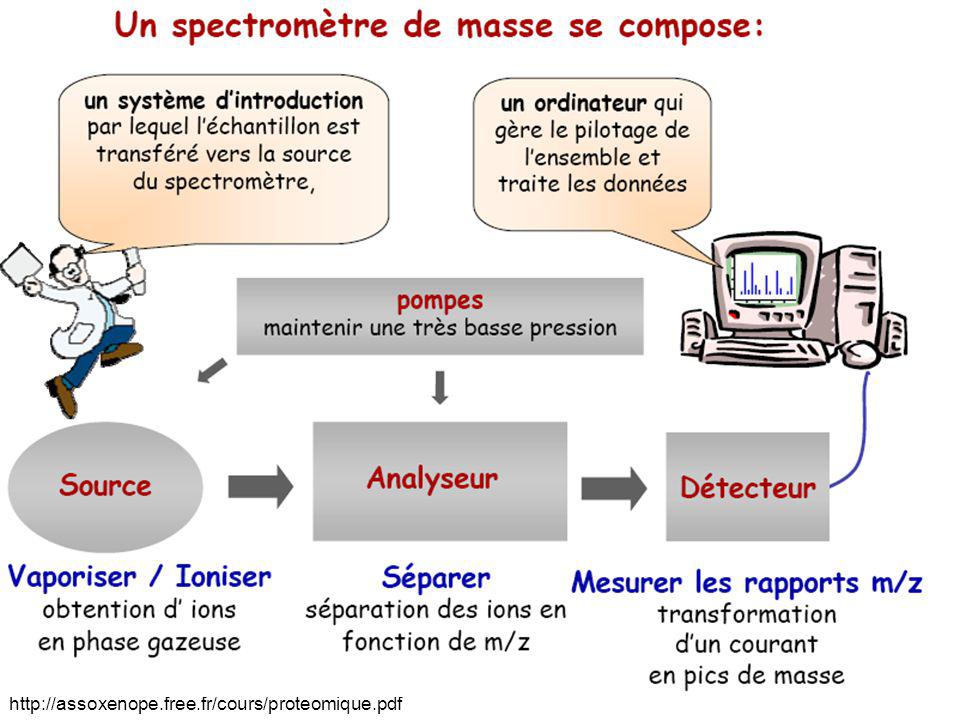
Détecteur en spectrométrie de masse
Chambre d'ionisation Jonction au silicium Scintillateur Cage de Faraday Détecteur à induction Multiplicateur d'électrons Détection dans un spectromètre de masse à résonance cylcotronique Détecteur hybride Détecteur cryogénique + grande diversité de fabricants Multitudes de types d’information et de formats de fichier

26
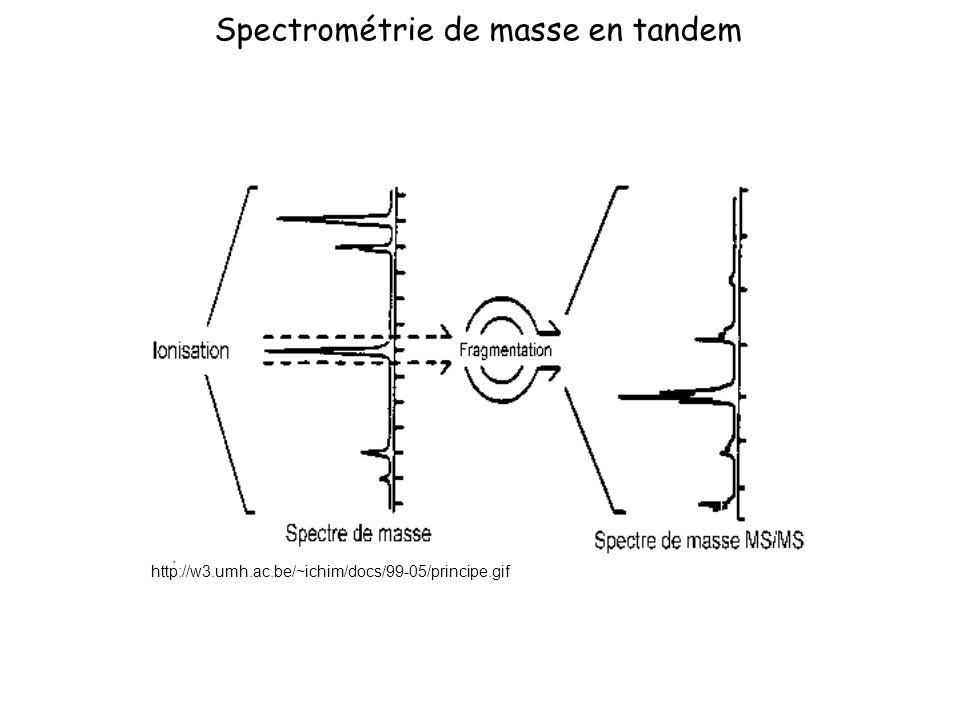
Spectrométrie de masse en tandem
27
Règles de fragmentation des peptides Biemann, 1990
2 a b c d R 1 R 3 CH 2 NH CH CO NH CH CO NH CH COOH 2 v w x y z Ions de série a, b et c : charge positive portée par la partie N-terminal Ions de série x, y et z : charge positive portée par la partie C-terminal Ions de série d, v et w : fragmentation des chaînes latérales Fragmentations basse énergie Fragmentations haute énergie Biemann K., Appendix 5, Nomenclature for peptide fragment ions (positive ions), Methods Enzymol, 1990, 193, 886-7 coursenligne.u-strasbg.fr/depotcel/DepotCel/279/Intranet/Carapito.ppt
, Methods Enzymol, 1990, 193, coursenligne.u-strasbg.fr/depotcel/DepotCel/279/Intranet/Carapito.ppt.")
28
Fragmentation des peptides : La loi du proton mobile
Dongré et al., Journal of Mass Spectrometry, Vol. 31, (1996) Les peptides ne cassent qu'une seule fois pour générer préférentiellement les fragments y et b Fragments b Fragments y +ALLLFSDGR+ +A LLLFSDGR+ +ALLLFSDGR+ Fragmentation dans la cellule de collision +AL LLFSDGR+ +ALLLFSDGR+ +ALL LFSDGR+ +ALLLFSDGR+ +ALLL FSDGR+ +ALLLF SDGR+ +ALLLFSDGR+ +ALLLFS DGR+ +ALLLFSDGR+ +ALLLFSD GR+ +ALLLFSDGR+ +ALLLFSDG R+ coursenligne.u-strasbg.fr/depotcel/DepotCel/279/Intranet/Carapito.ppt
 Les peptides ne cassent qu une seule fois pour générer préférentiellement les fragments y et b. Fragments b. Fragments y. +ALLLFSDGR+ +A. LLLFSDGR+ +ALLLFSDGR+ Fragmentation dans la cellule de collision. +AL. LLFSDGR+ +ALLLFSDGR+ +ALL. LFSDGR+ +ALLLFSDGR+ +ALLL. FSDGR+ +ALLLF. SDGR+ +ALLLFSDGR+ +ALLLFS. DGR+ +ALLLFSDGR+ +ALLLFSD. GR+ +ALLLFSDGR+ +ALLLFSDG. R+ coursenligne.u-strasbg.fr/depotcel/DepotCel/279/Intranet/Carapito.ppt.")
29
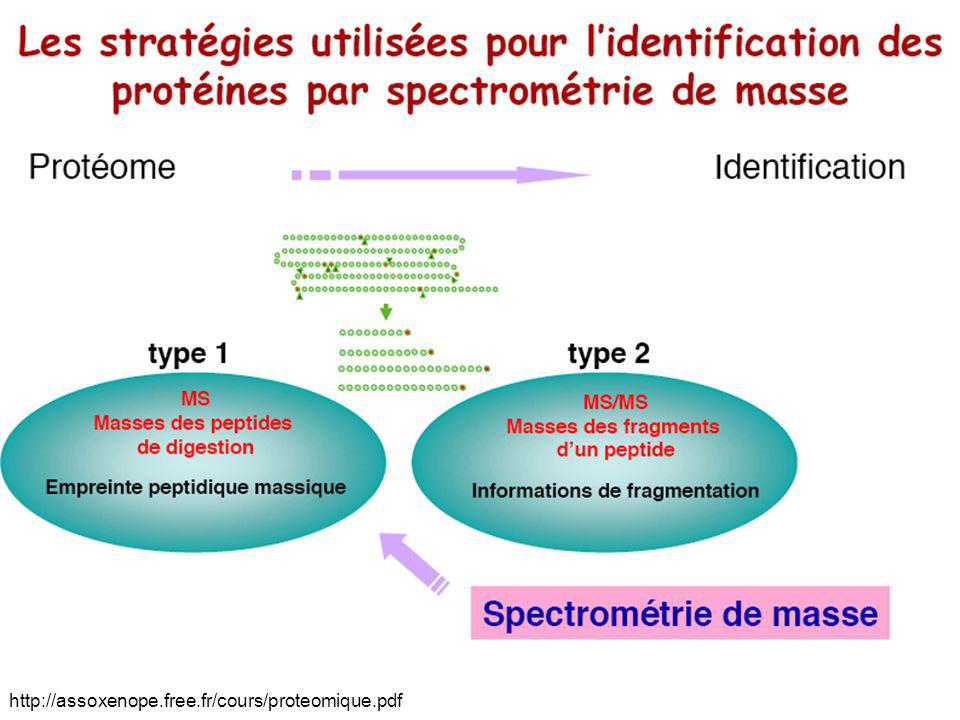
Exemple de spectre MS/MS
En abscisse, le rapport masse/charge ; en ordonnée, le pourcentage des ions possédant une masse donnée. En pratique, seul l'espacement entre les pics est interprété, pas leur hauteur. En interprétant ces espacements, il est possible de reconstituer la séquence peptidique. Sur cet exemple, la lecture du spectre de droite à gauche permet de reconstituer la séquence EWMPGQPR

31
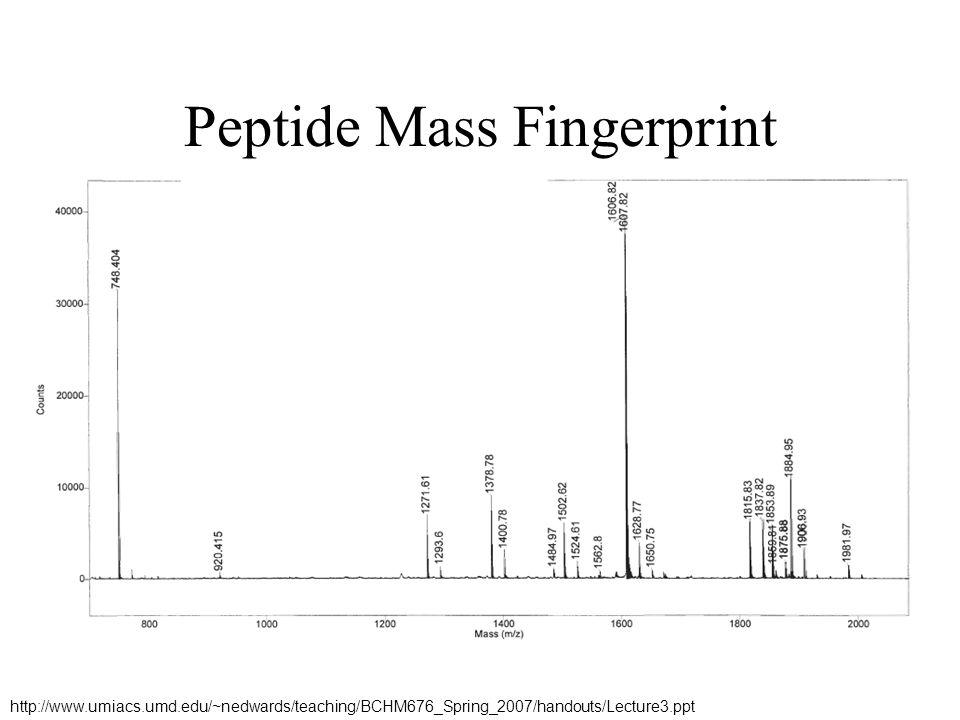
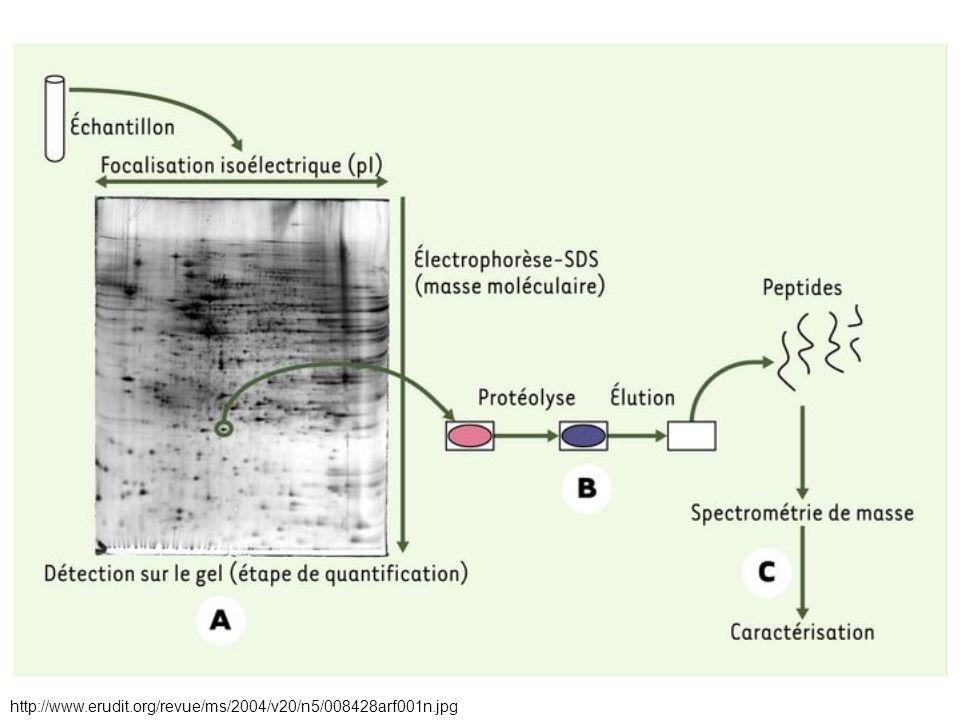
Peptide Mass Fingerprint
Concentrations as low as 10 femtomoles (10-15) Cut out 2D-Gel Spot
 Cut out. 2D-Gel Spot.")
32
Peptide Mass Fingerprint
Trypsin Digest

33
Peptide Mass Fingerprint
MS

34
Peptide Mass Fingerprint
35
Protein Sequence Myoglobin GLSDGEWQQV LNVWGKVEAD IAGHGQEVLI RLFTGHPETL EKFDKFKHLK TEAEMKASED LKKHGTVVLT ALGGILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISDA IIHVLHSKHP GDFGADAQGA MTKALELFRN DIAAKYKELG FQG

36
Protein Sequence Myoglobin GLSDGEWQQV LNVWGKVEAD IAGHGQEVLI RLFTGHPETL EKFDKFKHLK TEAEMKASED LKKHGTVVLT ALGGILKKKG HHEAELKPLA QSHATKHKIP IKYLEFISDA IIHVLHSKHP GDFGADAQGA MTKALELFRN DIAAKYKELG FQG

37
Peptide Masses 1811.90 GLSDGEWQQVLNVWGK 1606.85 VEADIAGHGQEVLIR
LFTGHPETLEK HGTVVLTALGGILK KGHHEAELKPLAQSHATK GHHEAELKPLAQSHATK YLEFISDAIIHVLHSK HPGDFGADAQGAMTK ALELFR

38
Peptide Mass Fingerprint
YLEFISDAIIHVLHSK GLSDGEWQQVLNVWGK GHHEAELKPLAQSHATK HGTVVLTALGGILK HPGDFGADAQGAMTK VEADIAGHGQEVLIR KGHHEAELKPLAQSHATK ALELFR LFTGHPETLEK

40
Sensitivité versus spécificité
PLOS Comp. Biol. (2008) 4:e12 Sensitivité : identifier le plus de protéines (le moins de faux négatifs) Spécificité : identifier le plus de vrais positifs
 4:e12. Sensitivité : identifier le plus de protéines (le moins de faux négatifs) Spécificité : identifier le plus de vrais positifs.")
41
Petit rappel Le Dalton est une unité de masse qui correspond à peu près à la masse d’un atome d’hydrogène. Exprimé en g, 1 Da correspond à environ 1, g. Glycine 75 Alanine 89 Sérine 105 Proline 115 Valine 117 Thréonine 119 Cystéine 121 Isoleucine 131 Leucine 131 Asparagine 132 Aspartate 133 Glutamine 146 Lysine 146 Glutamate 147 Méthionine 149 Histidine 155 Phénylalanine 165 Arginine 174 Tyrosine 181 Tryptophane 204 Précision de la spectrométrie de masse : 0,1 dalton à 10 daltons Modifications post-traductionnelles Acétylation 33 Méthylation 15 Glutamylation 146 Glycylation 75 Glycosylation >30 Isoprénylation >100 Phosphorylation 95

42
Digest with specific protease
Trypsin (K, R; not followed by P) Chymotrypsin (F, W, Y, L, M) Lys-C (K) Arg-C (R) Asp-N (D, N-terminal) V8-bicarb (E) V8-biphosph (E, D) {CNBr (M)} b
 Chymotrypsin (F, W, Y, L, M) Lys-C (K) Arg-C (R) Asp-N (D, N-terminal) V8-bicarb (E) V8-biphosph (E, D) {CNBr (M)} b.")
43
Digest with specific protease
Why trypsin? High specificity (K or R, not followed by P) Acetylated form commercially available (acetylation lessens autodigestion) Autolysis peaks are great internal calibrants ( and ) ( ), guanidinated
 Acetylated form commercially available. (acetylation lessens autodigestion) Autolysis peaks are great internal calibrants. ( and ) ( ), guanidinated.")
44
Digest with specific protease
546 aa 60 kDa; Da pI = 4.75 >RBME00320 Contig0311_ _ EC-mopA 60 KDa chaperonin GroEL MAAKDVKFGR TAREKMLRGV DILADAVKVT LGPKGRNVVI EKSFGAPRIT KDGVSVAKEV ELEDKFENMG AQMLREVASK TNDTAGDGTT TATVLGQAIV QEGAKAVAAG MNPMDLKRGI DLAVNEVVAE LLKKAKKINT SEEVAQVGTI SANGEAEIGK MIAEAMQKVG NEGVITVEEA KTAETELEVV EGMQFDRGYL SPYFVTNPEK MVADLEDAYI LLHEKKLSNL QALLPVLEAV VQTSKPLLII AEDVEGEALA TLVVNKLRGG LKIAAVKAPG FGDCRKAMLE DIAILTGGQV ISEDLGIKLE SVTLDMLGRA KKVSISKENT TIVDGAGQKA EIDARVGQIK QQIEETTSDY DREKLQERLA KLAGGVAVIR VGGATEVEVK EKKDRVDDAL NATRAAVEEG IVAGGGTALL RASTKITAKG VNADQEAGIN IVRRAIQAPA RQITTNAGEE ASVIVGKILE NTSETFGYNT ANGEYGDLIS LGIVDPVKVV RTALQNAASV AGLLITTEAM IAELPKKDAA PAGMPGGMGG MGGMDF

45
Digest with specific protease
Trypsin yields 47 peptides (theoretically) Peptide masses in Da:
 Peptide masses in Da:")
46
Digest with specific protease
Trypsin yields 47 peptides (theoretically) Peptide masses in Da:
 Peptide masses in Da:")
47
Théorie et Pratique Supposant que l’on connaisse la séquence exacte, on peut prédire une liste de peptides (et leur masse), mais on va en voir moins par MS Digestion incomplète : enzyme n’est pas parfait (e.g. trypsine coupe moins bien quand un a.a. basique est adjacent au site de clivage) empêchement stérique cinétique Peptides perdus au cours de l’expérience : lavages mauvaise ionisation Peptides supplémentaires : contaminations clivage non spécifique modifications (e.g. oxydation de la méthionine) Bruit dans les spectres est surtout chimique, pas electrique
, mais on va en voir moins par MS. Digestion incomplète : enzyme n’est pas parfait (e.g. trypsine coupe moins bien. quand un a.a. basique est adjacent au site de clivage) empêchement stérique. cinétique. Peptides perdus au cours de l’expérience : lavages. mauvaise ionisation. Peptides supplémentaires : contaminations. clivage non spécifique. modifications (e.g. oxydation de la méthionine) Bruit dans les spectres est surtout chimique, pas electrique.")
48
Digest with specific protease
Trypsin yields 47 peptides (theoretically) Peptide masses in Da:
 Peptide masses in Da:")
49
Seulement une protéine avec 5 matches parmi > 100 000 protéines
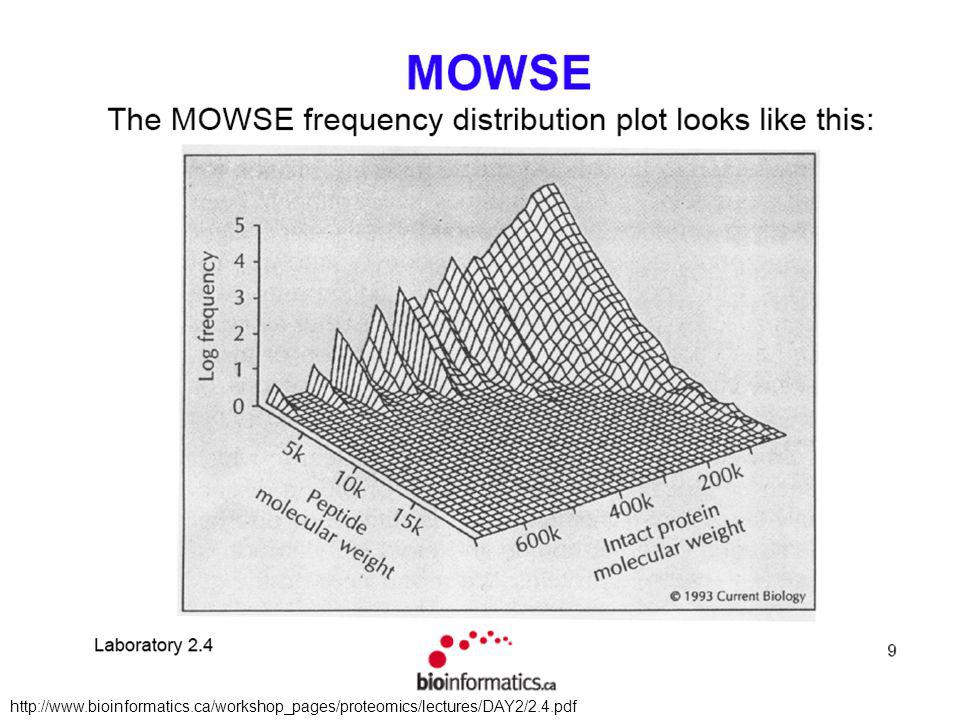
Fragfit : une approche simple Fragfit (PNAS, 1993, 90: Quel seuil choisir ? Favorise les grosses protéines (e.g. titine, 3 Mda) Requiert une liste de peptides (et leur masse) et une base de données de séquences protéiques Calcule, pour chaque protéine, la liste théorique Calcule, pour chaque protéine, le nombre de peptides qui “matchent” (en fonction d’un seuil défini a priori) Donne la liste des protéines par ordre décroissant de nombre de matches Remplacer le nombre de fragments trouvés par la fréquence des fragments trouvés Seulement une protéine avec 5 matches parmi > protéines Journal of the American Society for Mass Spectrometry (2003) 14:
 Requiert une liste de peptides (et leur masse) et une base de données de séquences protéiques. Calcule, pour chaque protéine, la liste théorique. Calcule, pour chaque protéine, le nombre de peptides qui matchent (en fonction d’un seuil défini a priori) Donne la liste des protéines par ordre décroissant de nombre de matches. Remplacer le nombre de fragments trouvés par la fréquence des fragments trouvés. Seulement une protéine avec 5 matches parmi > protéines. Journal of the American Society for Mass Spectrometry (2003) 14:")
50
http://www. bioinformatics

51
http://www. bioinformatics

52
http://www. bioinformatics

53
http://www. bioinformatics

54
http://www. bioinformatics
55
http://www. bioinformatics

56
http://www. bioinformatics

57
http://www. bioinformatics

58
http://www. bioinformatics

59
INDIQUER QUE L’ON PEUT ACCEPTER DES NON-COUPURES AVEC UNE CERTAINE FREQUENCE (CONTEXTE DEPENDANT)

60
MASCOT, une approche probabiliste
Perkins et al. (1999) Electrophoresis 20: Définir un modèle et calculer la probabilité qu’un spectre correspond à une protéine particulière Distribution a priori de la taille des peptides en fonction de la taille de la protéine Probabilité de non coupure par la protéase Probabilité de modifications post-traductionelles ou chimiques (en particulier des extrémités N et C terminales) Précision de la mesure de la masse (intervalle)
 Electrophoresis 20: Définir un modèle et calculer la probabilité qu’un spectre correspond à une protéine particulière. Distribution a priori de la taille des peptides en fonction de la taille de la protéine. Probabilité de non coupure par la protéase. Probabilité de modifications post-traductionelles ou chimiques (en particulier des extrémités N et C terminales) Précision de la mesure de la masse (intervalle)")
61
MASCOT, une approche probabiliste
Avantages : Fournit naturellement une e-value Corrige naturellement pour la taille des protéines Inconvénients : Temps calcul Limitations du modèle

62
Comparaison des cellules mutantes, cancéreuses, de deux écotypes différents, infectées par un virus, soumises à un médicament ou à un composé toxique Regarder les différentes protéines présentes, les modifications post-traductionnelles, les différents variants d’épissage ou d’édition Regarder les complexes protéiques RNAi

63
Génome versus Transcriptome et Protéome
Etude de la pleiotropie

64
Quelles améliorations ?
Fournir des informations supplémentaires (taille de la protéine, pI, etc.) Utiliser une banque de spectres pré-établis plutôt qu’une banque de séquences (suppose que le spectre est assez reproductible) Développer des statistiques permettant de garder le taux de faux positifs en dessous de e.g. 5% Filtrer les spectres de masse a priori pour éliminer les contaminants Utiliser des distributions plutôt que des intervalles pour prendre en compte l’erreur de mesure Identifier des mélanges de protéines (cadre Bayésien) Approche consensus Séquençage (étiquette - 3 a.a. - ou ensemble du fragment) Protéomique est une approche très séduisante. Mais le parallèle avec la génomique est trompeur. Le cadre théorique n’est pas le même : il y a 1 génome et une infinité de protéomes, car pour chaque gène, même chez les bactéries, il y a une population de séquences protéiques, qui diffèrent plus ou moins les unes des autres. Identifier le niveau d’expression du gène (avec toutes les difficultés que cela représente) ne permet pas de caractériser cette population. La base théorique de la protéomique est donc pour l’instant insuffisante et cela constituera une limite majeure à moyen terme. Cependant, à court terme, c’est une opportunité extraordinaire pour les biochimiques et pour les bio-informaticiens. La MS peut être ajoutée à la plupart des approches biochimiques existantes et fournir une meilleure, même si elle est imparfaite, caractérisation du protéome. Les bioinformaticiens peuvent améliorer les méthodes d’identification, même si ils sont souvent limités par la qualité des données (dépend des choix et des limites des méthodes biochimiques et physiques). Finalement, ne pas perdre de vue un effet néfaste de la protéomique (et de tout progrès scientifique en général), le coût environnemental est de plus en plus élevé : de plus en plus de machines, de plus en plus de données, donc plus de disques durs, plus d’ordinateurs, plus de temps calcul. Pour vous fixer les idées, dans un domaine de recherche tout à fait différent, la physique quantique. 1 million de dollars au Japon
 Utiliser une banque de spectres pré-établis plutôt qu’une banque de séquences (suppose que le spectre est assez reproductible) Développer des statistiques permettant de garder le taux de faux positifs en dessous de e.g. 5% Filtrer les spectres de masse a priori pour éliminer les contaminants. Utiliser des distributions plutôt que des intervalles pour prendre en compte l’erreur de mesure. Identifier des mélanges de protéines (cadre Bayésien) Approche consensus. Séquençage (étiquette - 3 a.a. - ou ensemble du fragment) Protéomique est une approche très séduisante. Mais le parallèle avec la génomique est trompeur. Le cadre théorique n’est pas le même : il y a 1 génome et une infinité de protéomes, car pour chaque gène, même chez les bactéries, il y a une population de séquences protéiques, qui diffèrent plus ou moins les unes des autres. Identifier le niveau d’expression du gène (avec toutes les difficultés que cela représente) ne permet pas de caractériser cette population. La base théorique de la protéomique est donc pour l’instant insuffisante et cela constituera une limite majeure à moyen terme. Cependant, à court terme, c’est une opportunité extraordinaire pour les biochimiques et pour les bio-informaticiens. La MS peut être ajoutée à la plupart des approches biochimiques existantes et fournir une meilleure, même si elle est imparfaite, caractérisation du protéome. Les bioinformaticiens peuvent améliorer les méthodes d’identification, même si ils sont souvent limités par la qualité des données (dépend des choix et des limites des méthodes biochimiques et physiques). Finalement, ne pas perdre de vue un effet néfaste de la protéomique (et de tout progrès scientifique en général), le coût environnemental est de plus en plus élevé : de plus en plus de machines, de plus en plus de données, donc plus de disques durs, plus d’ordinateurs, plus de temps calcul. Pour vous fixer les idées, dans un domaine de recherche tout à fait différent, la physique quantique. 1 million de dollars au Japon.")
Présentations similaires
























:>")