Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Analyse statistique des séquences génomiques
DEA en bioinformatique Lausanne, 3 mai 2001 Laurent Duret

2
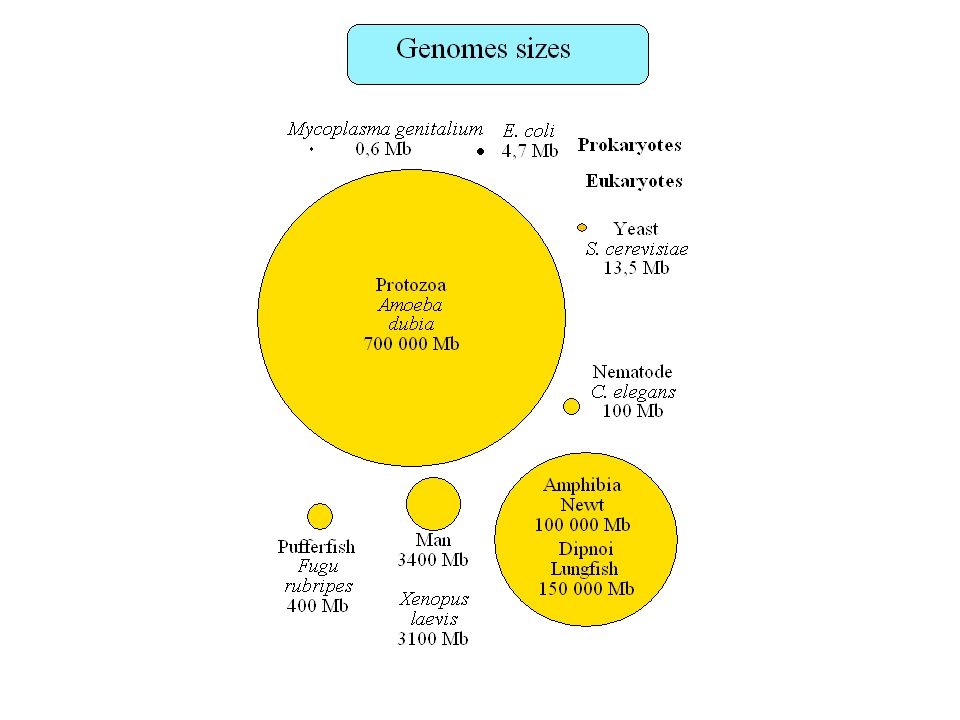
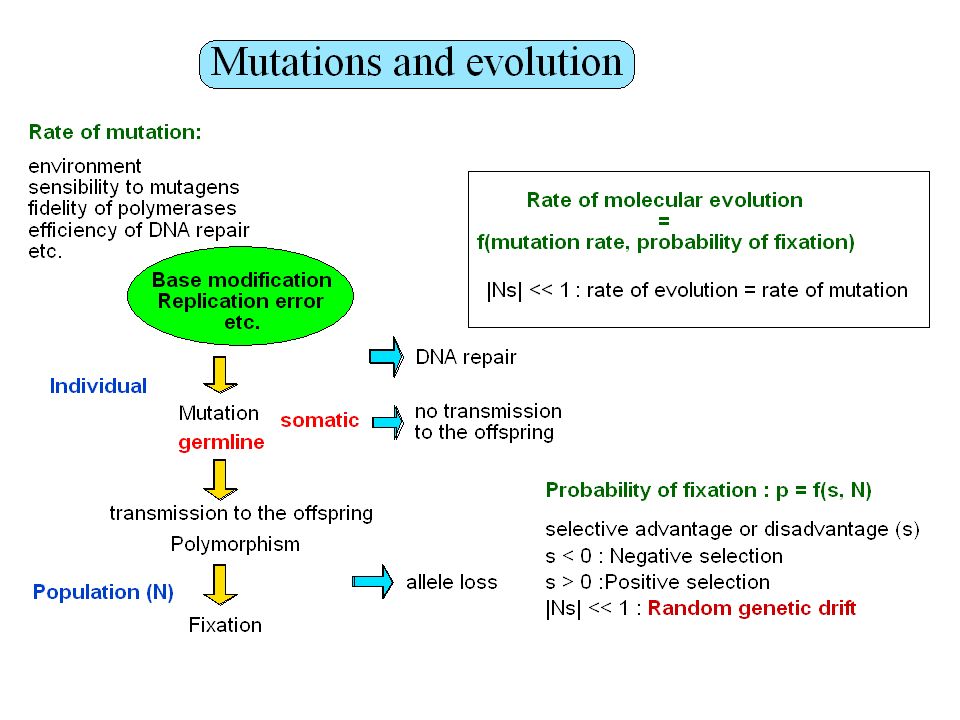
Plan Taille des génomes, paradoxe de la valeur C
Contenu informationnel Séquences répétées Organisation en isochore des génomes de vertébrés Prédiction de gène protéiques Prédiction de régions régulatrices non-codantes Usage des codons synonymes

7
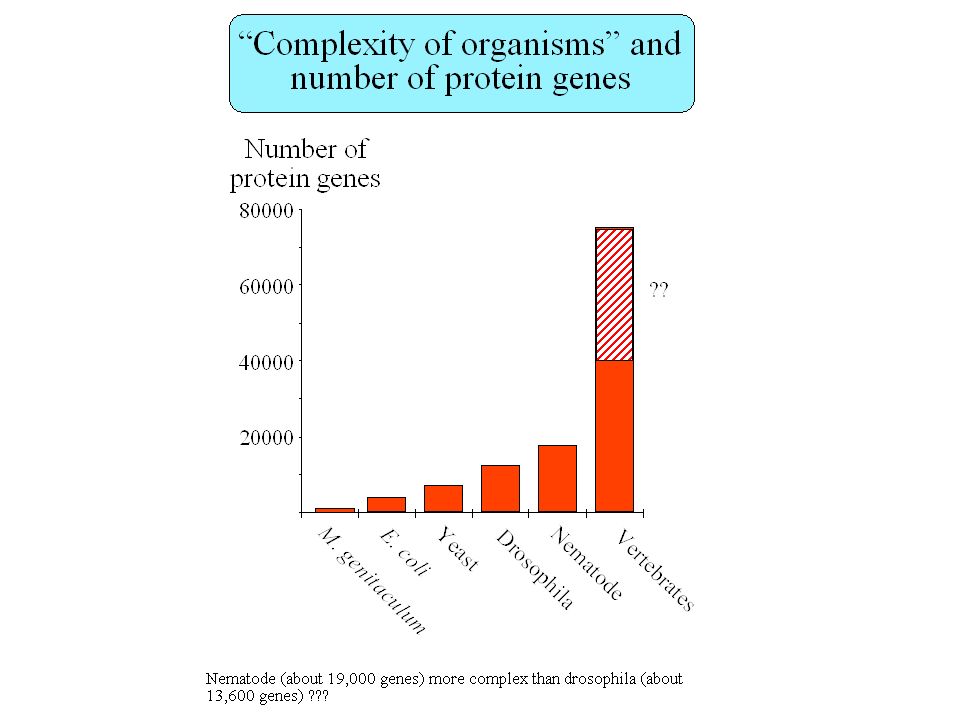
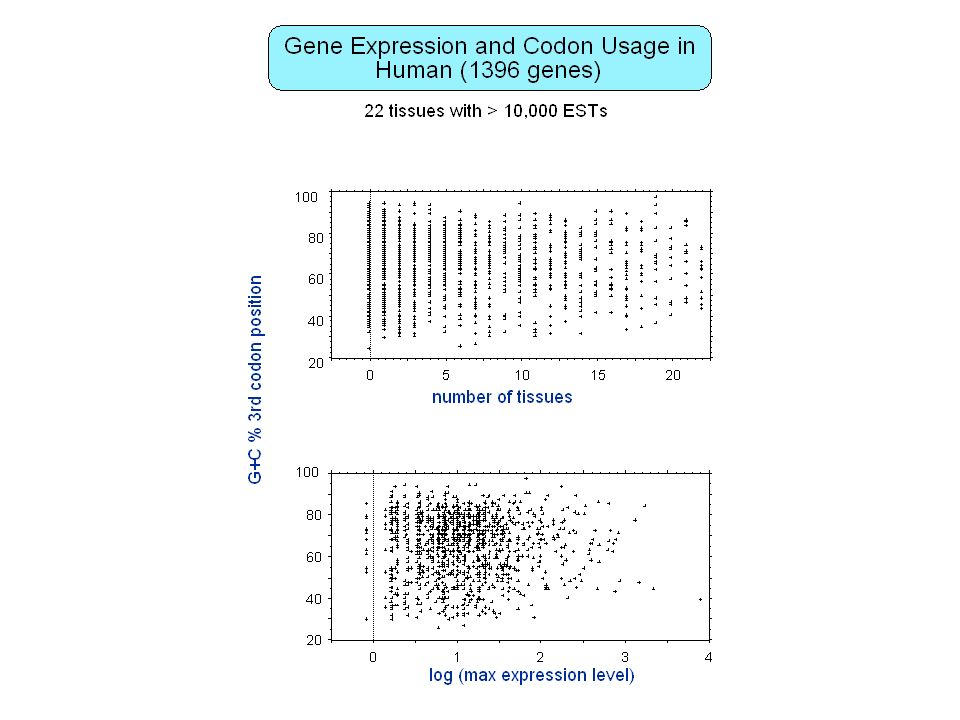
How many genes in the human genome ?

8
Functional elements in the human genome
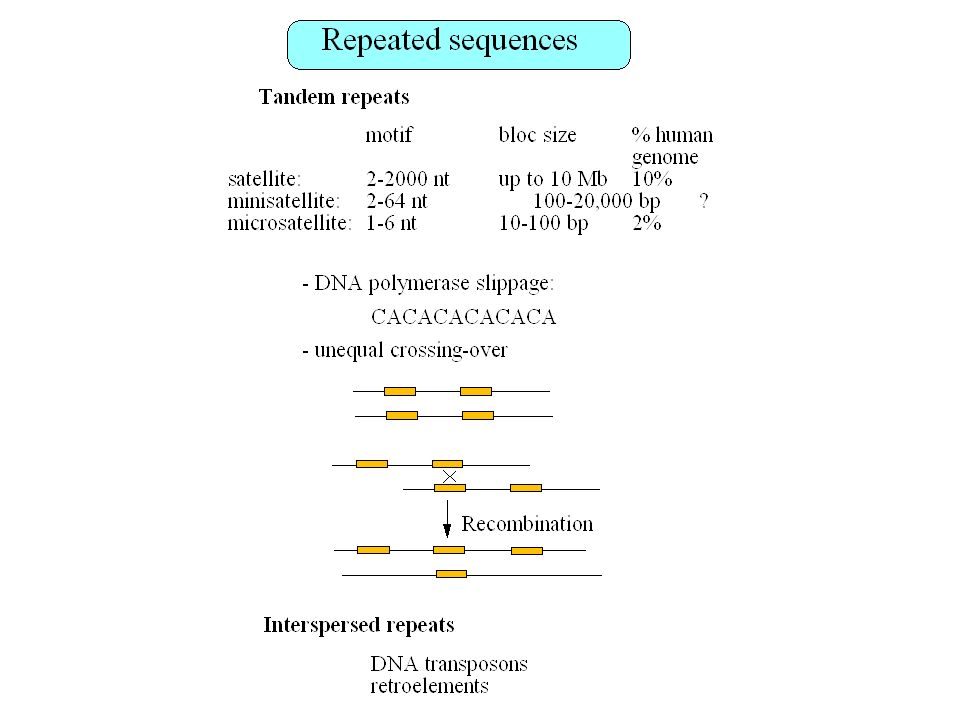
Untranslated RNAs: Xist, H19, His-1, bic, etc. Regulatory elements: promoters, enhancers, etc. Repeated sequences (SINES, LINES, HERV, etc.) : 40% of the human genome
 : 40% of the human genome.")
9
Structure of human protein genes
1396 complete human genes (exons + introns) from GenBank (1999) Average size (25%, 75%) Gene 15 kb ± 23 kb (4, 16) (10% > 35 kb) CDS nt ± 1200 (600, 1500) Exon (coding) 200 nt ± 180 (110, 200) Intron nt ± 3000 (500, 2000) 5'UTR 210 nt (Pesole et al. 1999) 3'UTR 740 nt (Pesole et al. 1999) Intron/exon Number of introns: 6 ±3 introns / kb CDS Introns / (introns + CDS): 80% 5' introns in 15% of genes (more ?), 3 ’introns very rare Alternative splicing in more than 30% of human genes (Hanke et al. 1999)
 from GenBank (1999) Average size (25%, 75%) Gene 15 kb ± 23 kb (4, 16) (10% > 35 kb) CDS 1300 nt ± 1200 (600, 1500) Exon (coding) 200 nt ± 180 (110, 200) Intron 1800 nt ± 3000 (500, 2000) 5 UTR 210 nt (Pesole et al. 1999) 3 UTR 740 nt (Pesole et al. 1999) Intron/exon. Number of introns: 6 ±3 introns / kb CDS. Introns / (introns + CDS): 80% 5 introns in 15% of genes (more ), 3 ’introns very rare. Alternative splicing in more than 30% of human genes (Hanke et al. 1999)")
10
Structure of human protein genes
GenBank: bias towards short genes 2408 complete human genes (exons + introns)
")
11
Structure of human protein genes
GenBank: bias towards short genes 1396 complete human genes (exons + introns) 9268 complete human mRNA
 9268 complete human mRNA.")
14
ADN satellite: centromères

18
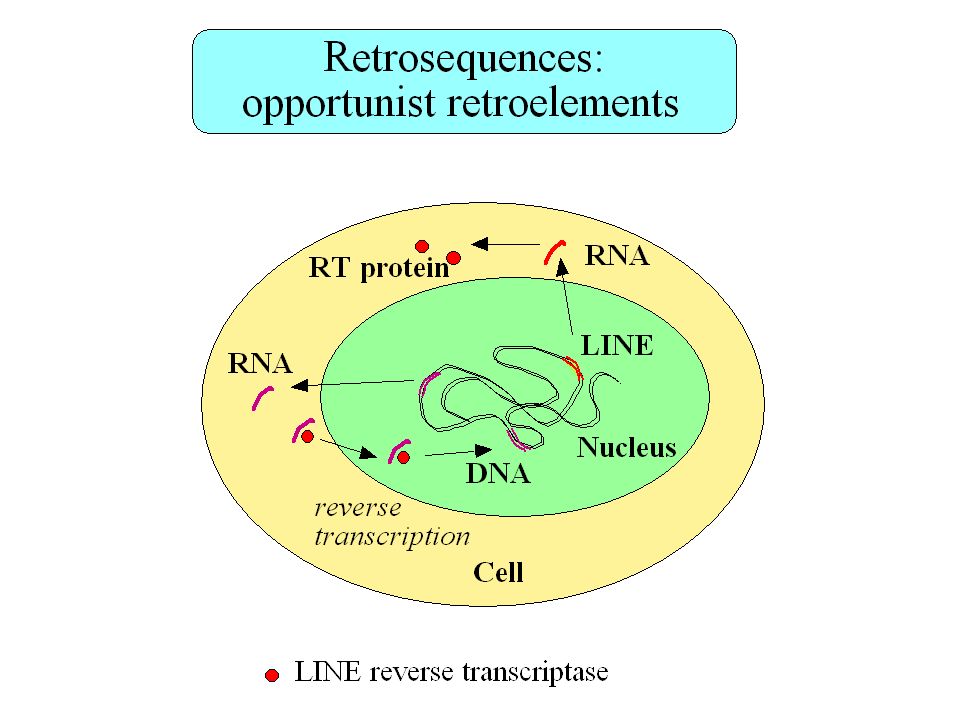
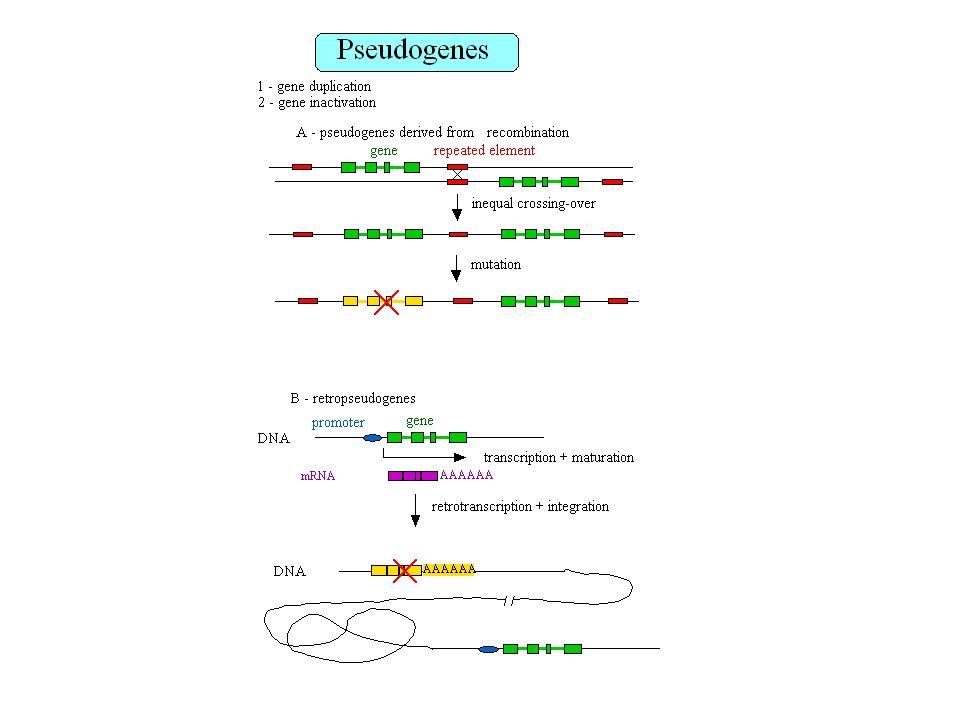
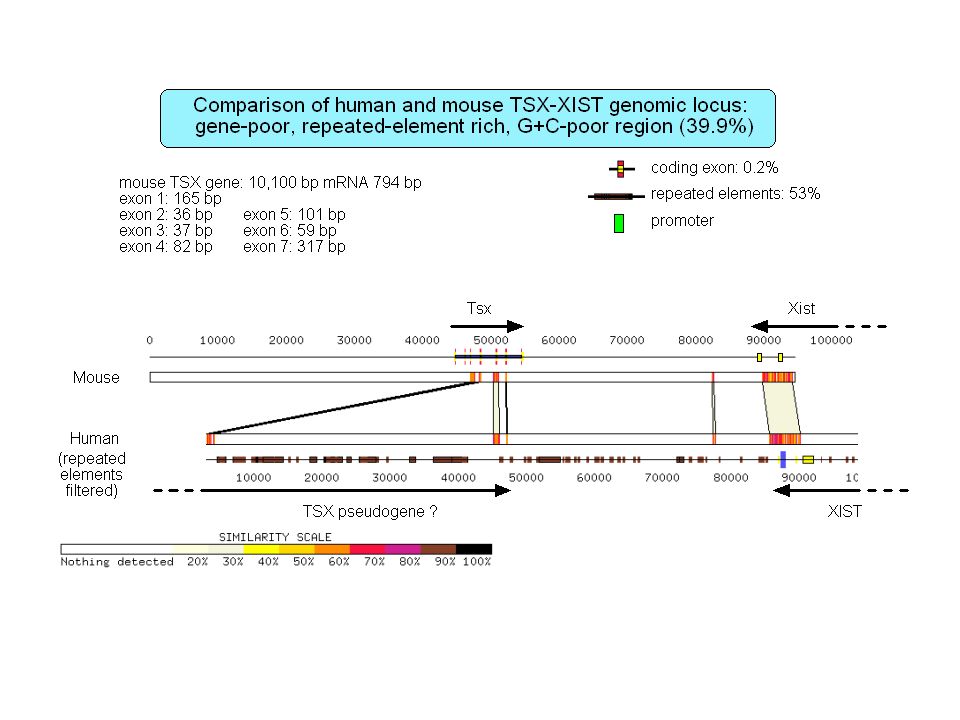
Retropseudogènes 23,000 à 33,000 retropseudogènes dans le génome humain Les gènes qui génèrent des retropseudogènes sont généralement de type housekeeping Gonçalves et al. 2000

19
Fréquence des éléments transposables dans le génome humain
Total = 42% (Smit 1999)
")
20
Fréquence des éléments transposables dans le génome humain (Smit 1999)
")
21
Isochore organization of vertebrate genomes

22
Organisation en isochore des génomes de vertébrés: mise en évidence expérimentale
Fractionnement du génome de la souris par centrifugation en gradient de densité (Bernardi et al. 1976)
")
23
Analyse statistique des séquences publiées dans les banques de données
Analyse statistique des séquences publiées dans les banques de données. Corrélation entre la composition en base en position 3 des codons et celle de l'envirronement génomique dans lequel se trouve le gène

24
Analyse statistique des séquences publiées dans les banques de données
Analyse statistique des séquences publiées dans les banques de données. Distribution en fréquence des gènes dans les différentes classes d'isochores 14 12 Moy = .580 Moy = .509 12 Ecart-t = .106 Ecart-t = .103 10 703 séq 10 173 séq 8 8 6 6 4 4 2 2 20 40 60 80 100 20 40 60 80 100 Nb de gènes (%) Danio Xénope 1 2 3 4 5 6 7 20 40 60 80 100 Moy = .612 Ecart-t = .158 5447 séq Moy = .639 Ecart-t = .171 818 séq Homme Poulet CDS GC3%
 Danio. Xénope Moy = Ecart-t = séq. Moy = Ecart-t = séq. Homme. Poulet. CDS GC3%")
25
Evolution de la structure en isochore chez les vertébrés

26
Isochore organization of vertebrate genomes
Insertion of repeated sequences (A. Smit 1996) Recombination frequency (Eyre-Walker 1993) Chromosome banding (Saccone, 1993) Replication timing (Bernardi, 1998) Gene density (Mouchiroud, 1991) Gene expression ?? -> No Gene structure (Duret, 1995)
 Recombination frequency (Eyre-Walker 1993) Chromosome banding (Saccone, 1993) Replication timing (Bernardi, 1998) Gene density (Mouchiroud, 1991) Gene expression -> No. Gene structure (Duret, 1995)")
27
Isochores and insertion of repeat sequences (Smit 1999)
4419 human genomic sequences > 50 kb

28
Isochores and gene density
MHC locus (3.6 Mb) (The MHC sequencing consortium 1999) Class I, class II (H1-H2 isochores): 20 genes/Mb, many pseudogenes Class III (H3 isochore): 84 genes/Mb, no pseudogene Class II boundaries correlate with switching of replication timing
 (The MHC sequencing consortium 1999) Class I, class II (H1-H2 isochores): 20 genes/Mb, many pseudogenes. Class III (H3 isochore): 84 genes/Mb, no pseudogene. Class II boundaries correlate with switching of replication timing.")
29
Isochores and introns length
Duret, Mouchiroud and Gautier, 1995 760 complete human genes L1L2: intron G+C content < 46% H1H2: intron G+C content 46-54% H3: intron G+C content >54%

30
Next steps in genome projects
Identify genes and other functional elements within genomic sequence (where are the genes ?) Determine the function of genes (what do they do ?)
 Determine the function of genes (what do they do )")
31
Prédiction ab initio de gènes eucaryotes
Introns, exons Prédiction de gènes protéiques Recherche de phases ouvertes de lecture (ORF: open reading frame) Phase +0 Phase +1 Phase +2 ATGTACCGTCGATCGTAGCTTGATCGATCG TACATGGCAGCTAGCATCGAACTAGCTAGC Phase -0 Phase -1 Phase -2 Taille moyenne des ORF: ± 150 nt Statistiques sur les nucléotides, usage des codons Hexamères Signaux d ’épissage
 Phase +0. Phase +1. Phase +2. ATGTACCGTCGATCGTAGCTTGATCGATCG. TACATGGCAGCTAGCATCGAACTAGCTAGC. Phase -0. Phase -1. Phase -2. Taille moyenne des ORF: ± 150 nt. Statistiques sur les nucléotides, usage des codons. Hexamères. Signaux d ’épissage.")
32
Prédiction de gènes eucaryotes (suite)
Construction d ’un modèle de gène protéique Combinaison d ’exons de phases compatibles (pondération en fonction des scores de chaque exon potentiel) Recherche de limites de gènes Exons terminaux (5 ’, 3 ’) Promoteur Signal de polyadénylation
 Recherche de limites de gènes. Exons terminaux (5 ’, 3 ’) Promoteur. Signal de polyadénylation.")
33
Prédiction de gènes eucaryotes: qualité de la prédiction
Comparaison des différents logiciels: sensibilité/spécificité Sensibilité : fraction des exons présents dans la séquence qui sont bien retrouvés Spécificité : fraction des vrais exons parmi tous ceux qui sont prédits Sn: sensibilité Sp: spécificité par exon (sn_e, sp_e) ou par nucéotide (sn_e, sp_e) Jeu de données Burset-Guigo (1996): 570 gènes de vertébrés
 ou par nucéotide (sn_e, sp_e) Jeu de données Burset-Guigo (1996): 570 gènes de vertébrés.")
34
Prédiction de gènes eucaryotes: qualité de la prédiction
Comparaison des différents logiciels: sensibilité/spécificité Sn: sensibilité Sp: spécificité par exon (sn_e, sp_e) ou par nucéotide (sn_e, sp_e) Locus BRCA2 (1.4 Mb, chrom. 13q) (Sanger Centre 1999): région "difficile" pour les logiciels de prédiction. 159 exons
 ou par nucéotide (sn_e, sp_e) Locus BRCA2 (1.4 Mb, chrom. 13q) (Sanger Centre 1999): région difficile pour les logiciels de prédiction. 159 exons.")
35
Prédiction de gènes protéiques complets
C. elegans: la plupart des ‘ gènes ’ annotés sont seulement des prédictions Peut-on utiliser ces méthodes pour annoter les séquences génomique humaines ? + les faux positifs ! + épissage alternatif ! exons non-codants !

36
Un peu d ’optimisme Fraction de la longueur des gènes correctement prédits: 70-80% Probabilité que deux exons potentiels consécutifs soient réels (et donc positifs en RT-PCR) 0.5
 0.5.")
37
Prediction of functional elements (2)
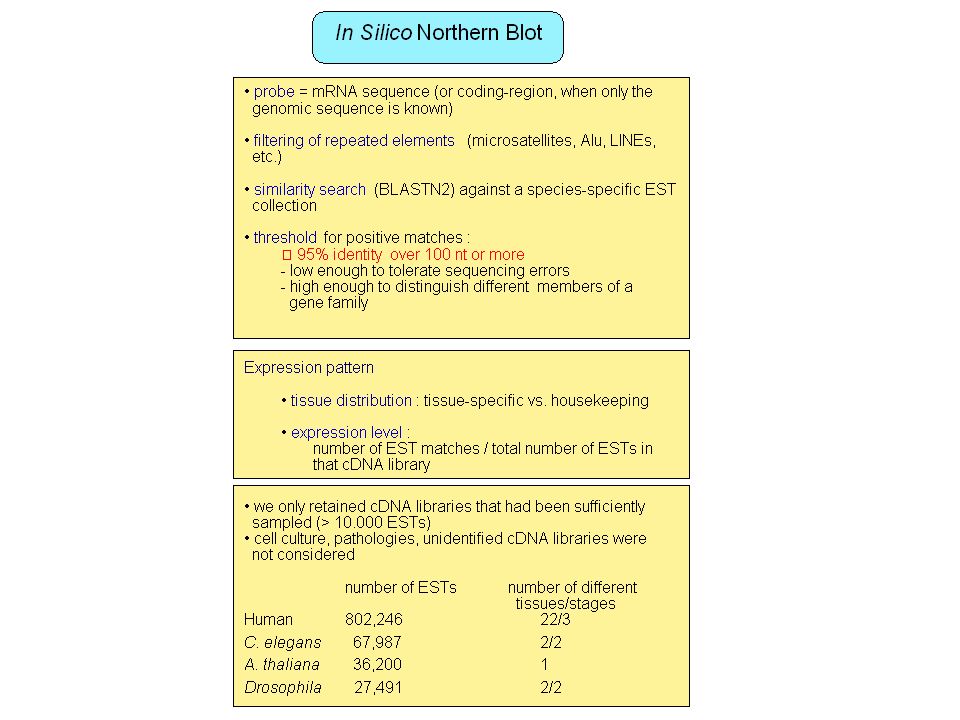
Large scale transcriptome projects: ESTs, full-length cDNA Identification of transcribed genes (protein or non-coding RNA) Information on alternative splicing, polyadenylation (Hanke et al. 1999, Gautheret et al. 1998), expression pattern SIM4: align a cDNA to genomic DNA Very useful but ... Problems with genes expressed at low level, narrow tissue distribution, stage-specific expression, … Limited tissue sampling Artifacts in ESTs (introns, partially matured RNA, …) Limited to polyadenylated RNA
 Information on alternative splicing, polyadenylation (Hanke et al. 1999, Gautheret et al. 1998), expression pattern. SIM4: align a cDNA to genomic DNA. Very useful but ... Problems with genes expressed at low level, narrow tissue distribution, stage-specific expression, … Limited tissue sampling. Artifacts in ESTs (introns, partially matured RNA, …) Limited to polyadenylated RNA.")
38
Prediction of functional elements (3)
Comparative sequence analysis (phylogenetic footprinting) Function => selective pressure Corollary Sequence conservation = selective pressure = function provided the number of aligned homologous sequences represents enough evolutionary time for the accumulation of mutations at the less constrained (presumably selectively neutral) base positions. Evolutionary rate in non-functional DNA: ~ 0.3% / My (± 0.069) Man/Mouse: ~ 80 Myrs 46-58% identity Mammals/Birds: ~ 300 Myr 26-28% identity Random sequences 25% identity
 Function => selective pressure. Corollary. Sequence conservation = selective pressure = function. provided the number of aligned homologous sequences represents enough evolutionary time for the accumulation of mutations at the less constrained (presumably selectively neutral) base positions. Evolutionary rate in non-functional DNA: ~ 0.3% / My (± 0.069) Man/Mouse: ~ 80 Myrs 46-58% identity. Mammals/Birds: ~ 300 Myr 26-28% identity. Random sequences 25% identity.")
39
Analyse comparative des gènes de b-actine de l'homme et de la carpe

42
Approche comparative (suite)
Comparaison d ’une séquence génomique avec des gènes déjà caractérisés dans d ’autres espèces (WISE2: alignement ADN/protéine avec épissage) Comparaison de séquences génomiques (non-annotées) homologues Locus mnd2 (homme souris) (Jang et al. 1999): >80 kb Prédiction d ’exons internes basée sur la conservation de séquence ORF ≥ 80 nt Séquence protéique ≥ 70% similarité Séquence ADN ≥50% identité GT AG conservés => détection de tous les exons internes du gène D6Mm5e Généralisation de la méthode : Sensibilité ? Spécificité ?
 Comparaison de séquences génomiques (non-annotées) homologues. Locus mnd2 (homme souris) (Jang et al. 1999): >80 kb. Prédiction d ’exons internes basée sur la conservation de séquence. ORF ≥ 80 nt. Séquence protéique ≥ 70% similarité. Séquence ADN ≥50% identité. GT AG conservés. => détection de tous les exons internes du gène D6Mm5e. Généralisation de la méthode : Sensibilité Spécificité")
43
Approche comparative: quelle est la bonne distance ? Guigo, Duret, Wiehe
Recherche d’exons codants conservés entre séquences génomiques orthologues (TBLASTX) 0.96 0.48 Homme/drosophile (670 Ma) 33 0.88 0.81 Homme/fugu (450 Ma) 0.87 0.85 Homme/poulet (300 Ma) 0.68 0.95 Homme/souris ( Ma) 0.53 0.99 Rat/souris (≈15 Ma) Sp_n Sn_n distance N
 Homme/drosophile (670 Ma) Homme/fugu (450 Ma) Homme/poulet (300 Ma) Homme/souris ( Ma) Rat/souris (≈15 Ma) 8. Sp_n. Sn_n. distance N.")
44
Phylogenetic footprinting
Advantages Works for all kinds of functional elements (transcribed or not, coding or not) as far as the information is in the primary sequence Does not require any a priori knowledge of the functional elements Limits Absence of evolutionary conservation does not mean absence of function No efficient method to detect unknown conserved secondary structure in RNA Function, but what function ? Number of sequences to compare : > 200 Myrs of evolution Mammals/birds: 310 Myrs Human + mouse + bovine : 240 Myrs
 as far as the information is in the primary sequence. Does not require any a priori knowledge of the functional elements. Limits. Absence of evolutionary conservation does not mean absence of function. No efficient method to detect unknown conserved secondary structure in RNA. Function, but what function Number of sequences to compare : > 200 Myrs of evolution. Mammals/birds: 310 Myrs. Human + mouse + bovine : 240 Myrs.")
45
Annotation systématique du génome humain
ENSEMBL project Human Genome Project Working Draft at UCSC The genome channel

46
Prédiction de régions régulatrices
Méthodes ab initio Prédiction de promoteurs Îlots CpG Approche comparative

47
Prédiction de promoteurs eucaryotes
Combinaison de sites de fixation de facteur de transcription (ordre, orientation, distance) Motifs courts, dégénérés Difficile de distinguer les vrais sites des faux positifs: Motif à 4 bases: ≈1/256 pb (1/128 pb sur les deux brins) Boîtes TATA, CAAT , GC: absents dans beaucoup de promoteurs Banques de données de sites de fixation de facteurs de transcription (TRANSFAC), de promoteurs caractérisés expérimentalement (EPD) PromoterScan (Prestridge 1995): Mesure de la densité en sites potentiels de fixation de facteurs de transcription de long de la séquence (pondération en fonction de la fréquence des sites dans ou en dehors des vrais promoteurs)
 Motifs courts, dégénérés. Difficile de distinguer les vrais sites des faux positifs: Motif à 4 bases: ≈1/256 pb (1/128 pb sur les deux brins) Boîtes TATA, CAAT , GC: absents dans beaucoup de promoteurs. Banques de données de sites de fixation de facteurs de transcription (TRANSFAC), de promoteurs caractérisés expérimentalement (EPD) PromoterScan (Prestridge 1995): Mesure de la densité en sites potentiels de fixation de facteurs de transcription de long de la séquence (pondération en fonction de la fréquence des sites dans ou en dehors des vrais promoteurs)")
48
Prédiction de promoteurs: sensibilité, spécificité
Sensibilité: fraction des promoteurs qui sont trouvés par le logiciel PromoterScan: sensibilité = 70% (promoteurs à boîte TATA) Spécificité: fraction des vrais promoteurs parmi ceux qui ont été prédits PromoterScan: spécificité = 20% Un faux positif / 10 kb Génome humain: ≈ gènes, ≈1 promoteur/30 kb
 Spécificité: fraction des vrais promoteurs parmi ceux qui ont été prédits. PromoterScan: spécificité = 20% Un faux positif / 10 kb. Génome humain: ≈ gènes, ≈1 promoteur/30 kb.")
49
Prédiction de promoteurs eucaryotes: recherches en cours
Prise en compte de l'orientation relative et des distances entre sites de fixation de facteurs de transcription COMPEL (Kolchanov 1998): banque de données d'éléments composites FastM : recherche dans une séquence génomique d'une combinaison de deux sites de fixation de facteurs de transcription à une distance définie l'un de l'autre Recherche de corrélations entre sites PromoterInspector (Werner 2000) Sensibilité: 40% Spécificité: 45% Combinaison recherche ab initio / approche comparative: recherche de sites potentiels parmi les régions conservées
: banque de données d éléments composites. FastM : recherche dans une séquence génomique d une combinaison de deux sites de fixation de facteurs de transcription à une distance définie l un de l autre. Recherche de corrélations entre sites. PromoterInspector (Werner 2000) Sensibilité: 40% Spécificité: 45% Combinaison recherche ab initio / approche comparative: recherche de sites potentiels parmi les régions conservées.")
50
Îlots CpG ou Génome de vertébrés : Me-C fortement mutable -> T
méthylation des C dans les dinucléotides 5 ’-CG-3 ’(CpG) Me-C fortement mutable -> T 5 ’-CG- 3 ’ 5 ’-TG-3 ’ ’-CA-3 ’ 3 ’-GC- 5 ’ ’-AC-5 ’ ’-GT-5 ’ Génome des vertébrés: globalement dépourvu en CpG (excès de TG, CA) Certaines régions (200 nt à plusieurs kb) échappent à la méthylation Pas de déplétion en CpG: CpGo/e proche de 1 Riche en G+C Îlot CpG: Longueur > 500 nt CpGo/e > 0.6 G+C > 50% ou
 Me-C fortement mutable -> T. 5 ’-CG- 3 ’ 5 ’-TG-3 ’ 5 ’-CA-3 ’ 3 ’-GC- 5 ’ 3 ’-AC-5 ’ 3 ’-GT-5 ’ Génome des vertébrés: globalement dépourvu en CpG (excès de TG, CA) Certaines régions (200 nt à plusieurs kb) échappent à la méthylation. Pas de déplétion en CpG: CpGo/e proche de 1. Riche en G+C. Îlot CpG: Longueur > 500 nt. CpGo/e > 0.6. G+C > 50% ou.")
51
La déamination des cytosines
CH H NH 2 O Uracile HN C CH N H O déamination réparation Cytosine N C CH H NH 2 O Cytosine méthylée N C CH H NH 2 O CH3 Thymine HN C CH N H O CH3 déamination TpG ou CpA

52
Îlots CpG: associé aux régions promotrices ?
Bird (1986), Gardiner-Garden (1987) Larsen (1992) ref 40% des gènes tissu-spécifiques possèdent un îlot CpG en 5 ’ 100% des gènes ‘ housekeeping ’ possèdent un îlot CpG en 5 ’ Rechercher des îlots CpG pour prédire des régions promotrices ? Sensibilité: % Spécificité ?? (Quelle fraction des îlots CpG correspond effectivement à des régions promotrices ?) Ponger (2001): comparaison des îlot CpG qui recouvre ou non le site d ’initiation de la transcription
, Gardiner-Garden (1987) Larsen (1992) ref. 40% des gènes tissu-spécifiques possèdent un îlot CpG en 5 ’ 100% des gènes ‘ housekeeping ’ possèdent un îlot CpG en 5 ’ Rechercher des îlots CpG pour prédire des régions promotrices Sensibilité: % Spécificité (Quelle fraction des îlots CpG correspond effectivement à des régions promotrices ) Ponger (2001): comparaison des îlot CpG qui recouvre ou non le site d ’initiation de la transcription.")
53
Fréquence des gènes humains avec un îlot CpG recouvrant le site d ’initiation de la transcription
800 gènes humains avec promoteur décrit Mesure de la distribution tissulaire à l ’aide d ’EST (20 tissus)
")
54
Comparaison des îlots CpG recouvrant ou non le site d ’initiation de la transcription
272 îlots start CpG recouvrant le site d ’initiation de la transcription (start) 1078 îlots CpG en dehors d ’un promoteur connu (other) (en excluant les séquences répétées)
 1078 îlots CpG en dehors d ’un promoteur connu (other) (en excluant les séquences répétées)")
55
Recherche de régions régulatrices par analyse comparative (empreintes phylogénétiques)
Goodman et al. 1988: régulation de l’expression des gènes du cluster b-globine au cours du développement Alignement de séquences orthologues de 6 mammifères (> 270 Ma d’évolution) 13 empreintes phylogénétiques: ≥ 6 nt, conservation 100% Analyse par retard de bande sur gel: 12/13 (92%) correspondent à des sites de fixation de protéines 1996: 35 empreintes phylogénétiques avec protéines fixatrices identifiées Enhancers de gènes HOX (Fugu/souris) (Aparicio et al. 1995) enhancer TCR a (homme/souris) (Luo, 1998) promoteur COX5B (11 primates) (Bachman, 1996) promoteur uPAR (homme/souris) (Soravia, 1995)
 13 empreintes phylogénétiques: ≥ 6 nt, conservation 100% Analyse par retard de bande sur gel: 12/13 (92%) correspondent à des sites de fixation de protéines. 1996: 35 empreintes phylogénétiques avec protéines fixatrices identifiées. Enhancers de gènes HOX (Fugu/souris) (Aparicio et al. 1995) enhancer TCR a (homme/souris) (Luo, 1998) promoteur COX5B (11 primates) (Bachman, 1996) promoteur uPAR (homme/souris) (Soravia, 1995)")
56
Large scale phylogenetic footprinting
Non-coding sequences : 325,247 sequences Mb everything except protein-coding regions and structural RNA genes (rRNA, tRNA, snRNA, scRNA) Introns, 5' and 3' untranslated regions, intergenic sequences Filtering of microsatellite repeats and cloning vectors: XBLAST Similarity search: BLASTN + LFASTA Vertebrates, insects, nematode
 Introns, 5 and 3 untranslated regions, intergenic sequences. Filtering of microsatellite repeats and cloning vectors: XBLAST. Similarity search: BLASTN + LFASTA. Vertebrates, insects, nematode.")
57
Metazoan Genome Projects

58
Sequence Similarities
1- Identification of new genes protein-genes, RNA-genes: intronic snoRNA genes 2- Retroviral elements, retrotransposons 3- Low complexity sequences: GC-rich, AT-rich, cryptic microsatellites 4- Artefacts: annotation errors, sample contamination (sponge insulin, ascidian RNA, chicken TGFB1) highly conserved regions (HCRs) - do not code for proteins - do not correspond to any known structural RNA
 highly conserved regions (HCRs) - do not code for proteins. - do not correspond to any known structural RNA.")
59
326 Highly Conserved Regions (HCRs)
• > 70% identity over 50 to 2000 nt after more than 300 Myrs • Unique sequences • Generally specific of only one gene • Longest HCR: 84% identity over 1930 nt after 300 Myrs 3’UTR deltaEF1 transcription factor • Oldest HCRs: 500 to 600 Myrs • No HCR between vertebrates and insects or nematode

60
Oldest HCRs

61
Conservation pattern in 3’UTRs

62
Distribution of HCRs within genes

63
HCRs and multigenic families

64
Function of 3’HCRs: mRNA stability, translation

65
Function of 3’HCRs: mRNA subcellular localization
Myosin heavy chain, c-myc, vimentin, b-actin

66
Comparaison des régions non-codantes de 77 gènes orthologues homme/souris (Jareborg et al. 1999)
Fraction des régions non-codantes conservées entre homme et souris

67
Next steps in genome projects
Identify genes and other functional elements within genomic sequence (where are the genes ?) Determine the function of genes (what do they do ?)
 Determine the function of genes (what do they do )")
68
Prédiction de fonction par homologie ?
Similarité entre séquences homologie Homologie structure conservée Structure conservée fonction conservée Oui, mais … Fonction: concept flou activité biochimique identique ? e.g. même ligand pour un récepteur, même substrat pour une enzyme, même gènes cibles pour un facteur de transcription. distribution tissulaire ? (isoformes tissu-spécifiques). compartimentalisation cellulaire: cytoplasme, mitochondrie, etc. Protéines homologues de fonction différentes Protéines homologues ligands (activateur/répresseur) d ’un même récepteur Recrutement pour une fonction totalement différente: t -cristalline / a-énolase Orthologie/paralogie Évolution modulaire
. compartimentalisation cellulaire: cytoplasme, mitochondrie, etc. Protéines homologues de fonction différentes. Protéines homologues ligands (activateur/répresseur) d ’un même récepteur. Recrutement pour une fonction totalement différente: t -cristalline / a-énolase. Orthologie/paralogie. Évolution modulaire.")
69
Prédiction de fonction par homologie ?
MZEORFG: 1 ILNSPDRACNLAKQAFDEAISELDSLGEESYKDSTLIMQLLXDNLTLWTSDTNEDGGDE 59 I N+P++AC LAKQAFD+AI+ELD+L E+SYKDSTLIMQLL DNLTLWTSD E BOV1433P: 186 IQNAPEQACLLAKQAFDDAIAELDTLNEDSYKDSTLIMQLLRDNLTLWTSDQQDEEAGE 244 Score = 87.4 bits (213), Expect = 1e-17 Identities = 41/59 (69%), Positives = 50/59 (84%) LOCUS BOV1433P bp mRNA MAM APR-1993 DEFINITION Bovine brain-specific protein eta chain mRNA, complete cds. ACCESSION J03868 LOCUS MZEORFG bp mRNA PLN MAY-1994 DEFINITION Zea mays putative brain specific protein, tau protein homolog mRNA, partial cds.
, Expect = 1e-17. Identities = 41/59 (69%), Positives = 50/59 (84%) LOCUS BOV1433P 1696 bp mRNA MAM 26-APR DEFINITION Bovine brain-specific protein eta chain mRNA, complete cds. ACCESSION J LOCUS MZEORFG 187 bp mRNA PLN 31-MAY DEFINITION Zea mays putative brain specific protein, tau protein. homolog mRNA, partial cds.")
70
Orthologie/paralogie

71
Approche phylogénétique pour la prédiction de fonction

72
Evolution modulaire

Présentations similaires
































 Phosphatases: retirent un phosphate.>")
 in mammalian genomes Organization of the human genome Human genome project: present status Human.>")
 in mammalian genomes Human genome sequence: 1300 Mb (38 %) available in GenBank (November 22 1999)>")