Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Les troubles de l’hémostase
M. Fontenay, Hôpital Cochin-SVP

2
Physiologie de l’hémostase

3
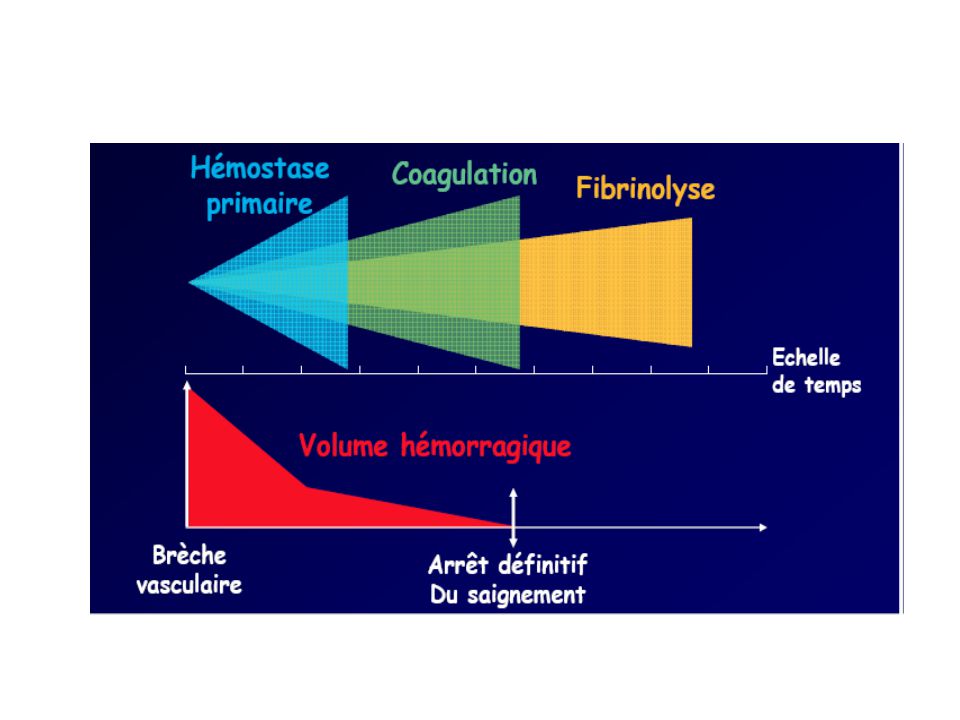
HEMOSTASE PRIMAIRE COAGULATION FIBRINOLYSE LESION VASCULAIRE
CAILLOT FIBRINO-PLAQUETTAIRE FIBRINOLYSE COLMATAGE DE LA BRECHE VASCULAIRE REPERMEATION DU VAISSEAU

4
Hémostase Coagulation Fibrinolyse primaire • Activation des facteurs •
Vasoconstriction de la coagulation minutes immédiate • Formation de fibrine • Lyse du caillot • Adhésion plaquettaire minutes heures secondes • Agrégation plaquettaire minutes

5
Endothélium vasculaire Facteurs plasmatiques : Facteurs plasmatiques
Hémostase Hémostase primaire Coagulation Endothélium vasculaire Plaquettes : glycoprotéines Facteurs plasmatiques : facteur Willebrand, fibrinogène Facteurs plasmatiques Plaquettes : membranes Caillot de fibrine Clou plaquettaire Caillot fibrino-plaquettaire

6
Hémostase primaire Vasoconstriction
3 étapes Vasoconstriction Adhésion des plaquettes au sous-endothélium Agrégation des plaquettes entre elles

7
Vasoconstriction

8
Adhésion des plaquettes au niveau de la brêche vasculaire
Agrégation des plaquettes Thrombus fibrino-plaquettaire

9
Plaquettes Cellules anucléées produites par les mégacaryocytes
150 à 450 G/L Durée de vie : 10 jours Structure Membrane : bicouche phospholipidique et glycoprotéines Cytosquelette Granules Denses (ADP, ATP, calcium, sérotonine Alpha : FP4, bTG, FV, PS, TGF-b, PDGF Système tubulaire dense (stockage du calcium intracellulaire)
")
11
Les différentes étapes de l’hémostase primaire
AGREGATION GPIIb-IIIa Fibrinogène Ca2+ ADP PF3 TxA2 SECRETION GPIb F. Willebrand ADHESION Endothélium Sous-endothélium

12
ENDOTHELIUM VASCULAIRE

13
Facteur von Willebrand
BRECHE VASCULAIRE Récepteur du Fg Plaquette Récepteur du FvW Facteur von Willebrand Collagène

14
BRECHE VASCULAIRE Plaquette vWF Récepteur du Fg Récepteur du FvW
Collagène

15
ADHESION PLAQUETTAIRE
Récepteur du Fg Plaquette Changement de forme vWF Récepteur du FvW Collagène

16
AGREGATION PLAQUETTAIRE
Récepteur du Fg Fibrinogène ADP Plaquette Plaquette activée TXA2 Récepteur du FvW vWF Collagène

17
Activation de la coagulation - génération de thrombine et formation de la fibrine
Fibrinogène FII FIIa Fibrine FIXa Ca2+ Ca2+ FVIIa FT Sous-endothelium

18
PK, KHPM, FXII, FXI, FIX / FVIII FT, FVIIa FX /FV Thrombine
VOIE DU CONTACT VOIE DU FACTEUR TISSULAIRE PL CA2+ PK, KHPM, FXII, FXI, FIX / FVIII FT, FVIIa FX /FV Thrombine Fibrinogène Fibrine

19
Syndrome hémorragique : Examen clinique et interrogatoire du patient
Définir le contexte : hémorragie isolée ou associée à maladie hépatique, rénale, infectieuse ? Type d’hémorragie : épistaxis, gingivorragies, hématomes, purpura… Evaluer le retentissement de l’hémorragie S’agit-il d’hémorragies spontanées ou provoquées ? Quels sont les médicaments pris par le patient ? Le patient a-t-il subi des interventions chirurgicales ou extractions dentaires suivies d’un saignement ? Antécédents hémorragiques personnels ou familiaux

20
Devant des signes hémorragiques
Numération plaquettaire (N : 150 – 450 x 109/L) Temps de saignement (TS) Duke à l’oreille (N < 3min mauvais) Ivy à l’avant-bras (N<10min) Test d’occlusion plaquettaire (TOP) Adrénaline (sérotonine) N < 145 sec ADP N<116 sec Attention : Hb < 8g/dL TS allongé Tests de coagulation (TQ, TCA) Dosages du fibrinogène et du facteur von Willebrand
 Temps de saignement (TS) Duke à l’oreille (N < 3min mauvais) Ivy à l’avant-bras (N<10min) Test d’occlusion plaquettaire (TOP) Adrénaline (sérotonine) N < 145 sec. ADP N<116 sec. Attention : Hb < 8g/dL TS allongé. Tests de coagulation (TQ, TCA) Dosages du fibrinogène et du facteur von Willebrand.")
21
Temps de Céphaline Activée (TCA) Temps de Quick (TQ)
VOIE DU CONTACT VOIE DU FACTEUR TISSULAIRE PL CA2+ PK, KHPM, FXII, FXI, FIX / FVIII FT, FVIIa FX /FV Thrombine Fibrinogène Fibrine

22
Protéines de la coagulation
1. Serine protéases a. Facteurs XI, XII b. Facteurs vitamine K-dépendants : Facteurs II, VII, IX, X Serine protéase II/IX IIa/IXa VII/X VIIa/Xa 2. Cofacteurs dépourvus d’activité enzymatique : FV, VIII V (proaccelerine) V’ Arg506 Arg306 VIII’ VIII (a-hemoph A)
 V’ Arg506. Arg306. VIII’ VIII (a-hemoph A)")
23
Exploration globale de la coagulation : TCA
Temps de céphaline activée (TCA): Réactif : céphaline = phospholipides Sensible à l’héparine et aux anticoagulants circulants de type lupique Explore : FXII, FXI, FX, FIX, FVIII, FII, Fg Surveillance des traitements par héparine standard Normes 34 sec +/- 5
: Réactif : céphaline = phospholipides. Sensible à l’héparine et aux anticoagulants circulants de type lupique. Explore : FXII, FXI, FX, FIX, FVIII, FII, Fg. Surveillance des traitements par héparine standard. Normes 34 sec +/- 5.")
24
Exploration globale de la coagulation : TQ
Temps de Quick (sec) : Taux de prothrombine (%) Réactif : Thromboplastine = facteur tissulaire Explore : FVII, FX, FV, FII, Fg Surveillance du traitement par antivitamine K Normes : 100% +/-25
 : Taux de prothrombine (%) Réactif : Thromboplastine = facteur tissulaire. Explore : FVII, FX, FV, FII, Fg. Surveillance du traitement par antivitamine K. Normes : 100% +/-25.")
25
Orientation diagnostique
Numération plaquettaire Thrombopénie < 50 x 109/L périphérique centrale Temps de saignement Normale ou augmentée Purpura vasculaire Normal TCA Allongé Thrombopathie Normal Willebrand Fg anormal Allongé

26
Eliminer une fausse thrombopénie sur EDTA

27
Fausse thrombopénie sur EDTA
Présence d’agrégats sur lame Contrôle de numération sur un tube citraté Si persistance d’agrégats faire : Soit un contrôle par prélèvement capillaire au bout du doigt Soit un contrôle sur héparine lithium (bouchon vert)
")
28
Mégacaryocytes absents Mécanisme périphérique
Mécanismes des thrombopénies Myélogramme Mégacaryocytes présents Mégacaryocytes absents Mécanisme périphérique Mécanisme central Destruction immunologique : - immunoallergique - autoimmune Destruction non immunologique : - Consommation : CIVD Pertes : Hémorragie massive - Séquestration : splénomégalie

29
Médicaments impliqués dans une thrombopénie immunoallergique

30
Purpura thrombopénique auto-immun
Thrombopénie avec ou sans signes hémorragiques cutanéo-muqueux Soit femme jeune (2/3 cas chronique) Soit enfant (transitoire, spontanément résolutif) Présence d’auto-anticorps anti-plaquettes (détectables dans 50% des cas) Lupus, HIV ou idiopathique Traitement : corticoïdes à fortes doses (prednisone à 1 mg/kg/jour pendant 15jours ou immunoglobulines intraveineuses (0.7 g/kg/j pendant 2 jours Si chronique supérieur à 3 mois : splénectomie
 Soit enfant (transitoire, spontanément résolutif) Présence d’auto-anticorps anti-plaquettes (détectables dans 50% des cas) Lupus, HIV ou idiopathique. Traitement : corticoïdes à fortes doses (prednisone à 1 mg/kg/jour pendant 15jours ou immunoglobulines intraveineuses (0.7 g/kg/j pendant 2 jours. Si chronique supérieur à 3 mois : splénectomie.")
31
Thrombopénies centrales
Absence de mégacaryocytes : Aplasie médullaire, insuffisance médullaire Toxicité : médicaments, alcool Envahissement de la moelle hémopathie : leucémie métastases Constitutionnelles rares

32
Thrombopathies - Principales causes
Définition: Allongement du TS avec numération plaquettaire normale Médicaments Inhibiteurs de cyclo-oxygénases : dérivés salicylés (ASPIRINE), anti-agrégants (PLAVIX, TICLID) et anti-inflammatoires inhibiteurs du recaptage de la sérotonine (antidépresseurs), antibiotiques (b-lactamine à fortes doses) Hémopathies syndromes myéloprolifératifs, syndromes myélodysplasiques, dysglobulinémies Autres pathologies insuffisance rénale, alcoolisme
, anti-agrégants (PLAVIX, TICLID) et anti-inflammatoires. inhibiteurs du recaptage de la sérotonine (antidépresseurs), antibiotiques (b-lactamine à fortes doses) Hémopathies. syndromes myéloprolifératifs, syndromes myélodysplasiques, dysglobulinémies. Autres pathologies. insuffisance rénale, alcoolisme.")
33
Orientation diagnostique
Numération plaquettaire Thrombopénie < 50 x 109/L périphérique centrale Temps de saignement Normale ou augmentée Purpura vasculaire Normal TCA Allongé Thrombopathie Normal Willebrand Fg anormal Allongé

34
Maladie de Willebrand Numération plaquettaire le plus souvent normale
Temps de saignement allongé, TCA allongé si FVIII inférieur à 50%

35
ADHESION PLAQUETTAIRE
GPIIbIIIa Plaquette Changement de forme GPIb vWF Collagène

36
Maladie de Willebrand Type 1 : déficit partiel autosomal dominant
FVIIIc : entre 20 et 50% (N>60%) Facteur Willebrand activité et antigène entre 20-50% Type 2 : déficit qualitatif FVIIIc souvent normal Facteur Willebrand antigène>activité Type 3 : déficit complet FVIIIc bas Facteur Willebrand antigène et activité < 10%
 Facteur Willebrand activité et antigène entre 20-50% Type 2 : déficit qualitatif. FVIIIc souvent normal. Facteur Willebrand antigène>activité. Type 3 : déficit complet. FVIIIc bas. Facteur Willebrand antigène et activité < 10%")
37
Willebrand type 1 Correction pendant la grossesse ou sous pilule
Hémorragies cuténeo-muqueuses : risque d’anémie par carence martiale Traitement : MINIRIN (desmopressine) à la dose de 0.3 µg/kg en perfusion lente de 20 min dans 50cc de serum physiologique Permet d’éviter les transfusions de culots globulaires
 à la dose de 0.3 µg/kg en perfusion lente de 20 min dans 50cc de serum physiologique. Permet d’éviter les transfusions de culots globulaires.")
38
Pathologie de la coagulation
Hémophilie

39
Temps de Céphaline Activée (TCA)
VOIE DU CONTACT VOIE DU FACTEUR TISSULAIRE PL CA2+ PK, KHPM, FXII, FXI, FIX / FVIII FT, FVIIa FX /FV Thrombine Fibrinogène Fibrine

40
Conduite à tenir devant un TCA allongé
Orientation clinique: antécédents personnels et familiaux d’hémorragie et prises médicamenteuses (héparine) TQ TQ (V,X,II) Atteinte hépatique Avitaminose K Déficit Constitutionnel TQ normal TCA M+T TCA M+T non corrigé Anticoagulant circulant ACC de type lupus Inhibiteur spécifique TCA M+T corrigé: Dosages des facteurs VIII: hémophilie A, Willebrand IX: hémophilie B XI XII: pas de risque hémorragique
 TQ. TQ (V,X,II) Atteinte hépatique. Avitaminose K. Déficit Constitutionnel. TQ normal. TCA M+T. TCA M+T non corrigé. Anticoagulant circulant. ACC de type lupus. Inhibiteur spécifique. TCA M+T corrigé: Dosages des facteurs. VIII: hémophilie A, Willebrand. IX: hémophilie B. XI. XII: pas de risque hémorragique.")
41
Causes d’un allongement du TCA
Traitement anticoagulant par héparine standard et certaines HBPM à durée de vie longue (Innohep, Fraxodi) Contamination involontaire d’un prélèvement par de l’héparine Déficit en facteur VIII, IX, XI ou XII Anticoagulant circulant
 Contamination involontaire d’un prélèvement par de l’héparine. Déficit en facteur VIII, IX, XI ou XII. Anticoagulant circulant.")
42
Hémophilie A ou B Hémorragies des séreuses
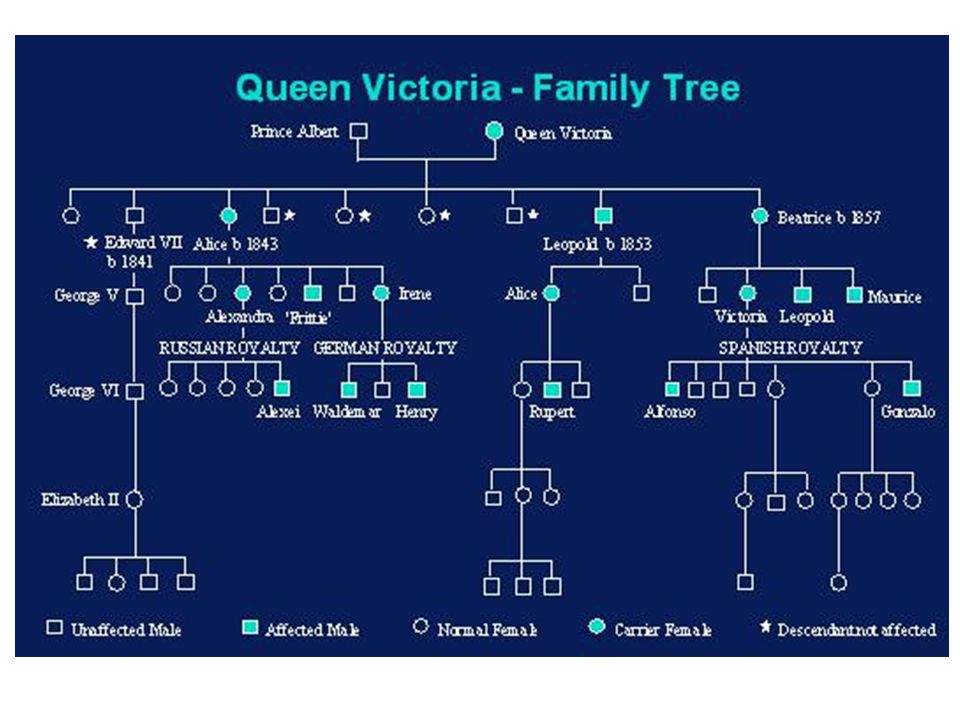
Hémophilie A : 1/ ; hémophilie B : 1/30 000 Transmission récessive liée à l’X Hémorragies des séreuses intra-articulaires, intramusculaires, intracrâniennes, urinaires Différents types sévère : < 1% modérée : 1% - 5% mineure : 5% - 50% Femmes conductrices à taux bas (15-50%) risque hémorragique + I II III Diagnostic prénatal dans les formes sévères - Possible dans 90% des cas
 risque hémorragique + I. II. III. Diagnostic prénatal dans les formes sévères. - Possible dans 90% des cas.")
44
Hématome musculaire

46
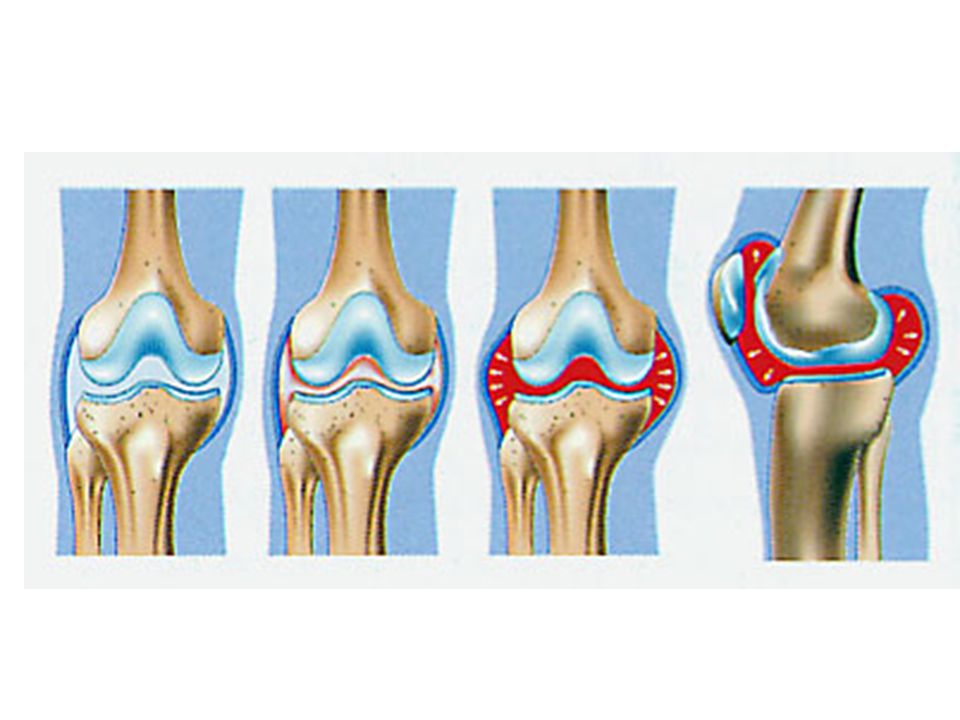
Arthropathie hémophilique du genou

47
CAT devant un sujet présentant une pathologie hémorragique de l’hémostase (hémophile)
Identification de l’anomalie/Certificat médical Ne pas faire de geste invasif sans traitement prophylactique CI des injections intra-musculaires Compression prolongée aux points de ponction Préserver le capital veineux périphérique Education des patients sur leur risque hémorragique Auto-perfusion

48
Conduite à tenir devant un TCA allongé
Orientation clinique: antécédents personnels et familiaux d’hémorragies, prises médicamenteuses (héparine) TQ TQ (V,X,II) Atteinte hépatique Avitaminose K Déficit Constitutionnel TQ normal TCA M+T TCA M+T non corrigé Anticoagulant circulant ACC de type lupus Inhibiteur spécifique TCA M+T corrigé: Dosages des facteurs VIII: hémophilie A, Willebrand IX: hémophilie B XI XII: pas de risque hémorragique
 TQ. TQ (V,X,II) Atteinte hépatique. Avitaminose K. Déficit Constitutionnel. TQ normal. TCA M+T. TCA M+T non corrigé. Anticoagulant circulant. ACC de type lupus. Inhibiteur spécifique. TCA M+T corrigé: Dosages des facteurs. VIII: hémophilie A, Willebrand. IX: hémophilie B. XI. XII: pas de risque hémorragique.")
49
TCA M+T allongé : Anticoagulant circulant
Allongement du TCA non corrigé par addition de plasma normal Si présence d’un anticoagulant circulant de type antiphospholipide Pas d’incidence hémorragique Incidence thrombotique pour certains types : anticardiolipine (ACL), anti-b2GP1 (thromboses artérielles ou veineuses et fausse-couches précoces)
, anti-b2GP1 (thromboses artérielles ou veineuses et fausse-couches précoces)")
50
TCA M+T allongé : Inhibiteurs anti-facteurs de la coagulation
Auto anti-FVIII : lupus, post-partum, hémopathies malignes, médicaments (sulfamides, phenylbutazone) : Syndrome hémorragique grave Allo-anticorps après transfusion de facteur VIII ou IX chez l’hémophile : Inefficacité des transfusions
 : Syndrome hémorragique grave. Allo-anticorps après transfusion de facteur VIII ou IX chez l’hémophile : Inefficacité des transfusions.")
51
Causes d’un allongement du TQ
TQ >12sec (TP<70%) Insuffisance hépato-cellulaire Déficit en vitamine K : diminution des facteurs K-vitamino-dépendants (II, VII, X, PC, PS) Traitement anti-vitamine K (Previscan, Sintrom, Coumadine) Déficit isolé constitutionnel en facteur VII, X, II, V CIVD Allongement isolé du TQ : déficit en facteur VII
 Insuffisance hépato-cellulaire. Déficit en vitamine K : diminution des facteurs K-vitamino-dépendants (II, VII, X, PC, PS) Traitement anti-vitamine K (Previscan, Sintrom, Coumadine) Déficit isolé constitutionnel en facteur VII, X, II, V. CIVD. Allongement isolé du TQ : déficit en facteur VII.")
52
Pathologie de la fibrinolyse
CIVD

53
Fibrinolyse Inhibiteur Activateur Plasmine Cell. endothéliale
Plasminogène Plasmine Fibrine D. Dimères

54
Exploration de la fibrinolyse
D-Dimères Protéolyse de la fibrine par la plasmine Présence de D-dimères = lyse d’un caillot de fibrine

55
D-Dimères

56
Coagulation IntraVasculaire Disséminée
Hémorragie secondaire à un phénomène de coagulation diffus dû à une génération de thrombine Fibrinolyse réactionnelle des caillots par la plasmine

57
Coagulation IntraVasculaire Disséminée
Bilan d’hémostase : D-dimères>50 µg/ml, thrombopénie sévère et aiguë, facteurs de la coagulation effondrés (V, VIII), Fibrinogène > 1g/l, complexes solubles positifs Plasminogène Plasmine Monomères de fibrine Complexes solubles Fibrine D-dimères IIa
, Fibrinogène > 1g/l, complexes solubles positifs. Plasminogène. Plasmine. Monomères. de fibrine. Complexes solubles. Fibrine. D-dimères. IIa.")
58
Causes de CIVD Hémorragie de la délivrance
Hématome rétro-placentaire pré-éclampsie Rétention d’œuf mort Placenta praevia Sepsis à bactérie gram-, viroses Cancer, leucémie, hémolyses intravasculaires, hypoxie, hypoperfusion

59
Diagnostic différentiel : Insuffisance hépato-cellulaire
Allongement du TQ et du TCA Défaut de synthèse des facteurs de la coagulation : FV, Fibrinogène Malabsorption de la vitamine K (cholestase) Persistance de précurseurs inactifs (FII, VII, IX, X) Anomalies qualitatives : hyperfibrinolyse (augmentation discrète des D-dimères) Thrombopénie : hypersplénisme
 Persistance de précurseurs inactifs (FII, VII, IX, X) Anomalies qualitatives : hyperfibrinolyse (augmentation discrète des D-dimères) Thrombopénie : hypersplénisme.")
60
Fibrinogénolyse primitive
Syndrome hémorragique du à une libération massive de plasmine par le placenta Lyse illégitime du fibrinogène indépendante de la formation de caillot Bilan d’hémostase : Fg>1g.L, peu de D-dimères, temps de lyse des euglobulines effondré < 10 min, thrombopénie modérée, diminution modérée des facteurs de la coagulation Fibrinogène Fibrine Produits de dégradation D-dimères Plasminogène Plasmine

61
Causes de la fibrinogénolyse primitive
Embolie amniotique (rare) : liquide amniotique riche en activateurs du plasminogène et diminution en fin de grossesse de l’inhibiteur de la plasmine, l’a2-antiplasmine. Cancer, leucémie, affections hépatiques sévères avec libération accrue d’activateurs du plasminogène (d’où génération de plasmine)
 : liquide amniotique riche en activateurs du plasminogène et diminution en fin de grossesse de l’inhibiteur de la plasmine, l’a2-antiplasmine. Cancer, leucémie, affections hépatiques sévères avec libération accrue d’activateurs du plasminogène (d’où génération de plasmine)")
62
+ tests FIBRINOLYSE PRIMITIVE CIVD Fg FV, FVIII FII FVII+X Plaquettes
D-Di CS Lyse N _ (<30min) + N
 + N.")
64
PK, KHPM, FXII, FXI, FIX / FVIII FT, FVIIa FV /FX Thrombine
VOIE DU CONTACT VOIE DU FACTEUR TISSULAIRE PL CA2+ PK, KHPM, FXII, FXI, FIX / FVIII FT, FVIIa FV /FX Antithrombine Protéines C et S Thrombine Fibrinogène Fibrine

65
Antithrombine Normes : synthèse hépatocytaire
Adulte : % Enfant : valeurs de l’adulte vers 1 an synthèse hépatocytaire Augmente dans les syndromes inflammatoires

66
Protéine C Adulte: % Enfant valeurs de l’adulte vers 15 ans

67
Protéine S Normales 2 formes: 40% libre, 60% liée à C4BbP.
H et F > 50 ans : % F < 50 ans : % 2 formes: 40% libre, 60% liée à C4BbP. Synthèse endothéliale et stockage plaquettaire, vitamino-K dpdt.

68
Autres causes génétiques d’hypercoagulabilité
Résistance du facteur V à l’inactivation par la protéine C activée Mutation G20210A du gène du facteur II (prothrombine) augmentation du taux de facteur II
 augmentation du taux de facteur II.")
69
Autres causes acquises d’hypercoagulabilité sanguine
Anticoagulant circulant (ACC), anticardiolipines (ACL) Thrombocytose
, anticardiolipines (ACL) Thrombocytose.")
70
Risque de thrombose Plaquettes F Willebrand Fibrinogène
FII, VII, IX, X FV, FVIII FXI, FXIII ACC, ACL Déficit en AntithrombineProtéine C ou Protéine S Ou RPCA Ou Mut FII

71
En pratique, Bilan d’hémostase Qualité du prélèvement
Tube citraté (bouchon bleu) Prélevé après élimination des premiers cc de sang Biologie sur plasma citraté après centrifugation Test d’occlusion sur sang total à préciser Numération plaquettaire sur EDTA
 Prélevé après élimination des premiers cc de sang. Biologie sur plasma citraté après centrifugation. Test d’occlusion sur sang total à préciser. Numération plaquettaire sur EDTA.")
Présentations similaires










>")








