Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Pathologie des troubles du mouvement et des muscles
Unité III – Neurologie 2016

2
Objectifs Décrire la neuropathologie des maladies suivantes :
Maladie de Parkinson Maladie de Huntington Paralysie supranucléaire progressive Sclérose latérale amyotrophique Reconnaître l’aspect histopathologique des muscles dans les cas suivants : Dénervation et réinnervation Myopathies inflammatoires Dystrophie musculaire

3
Divulgation Vous pouvez accéder et utiliser cette présentation PowerPoint à des fins éducatives seulement. Il est strictement défendu d’afficher cette présentation en ligne ou de la distribuer sans l’autorisation de l’auteur.

4
Troubles du mouvement Maladies de surcharge des protéines :
Surcharge de protéines tau : paralysie supranucléaire progressive Synucléine : maladie de Parkinson Huntingtine : maladie de Huntington Ubiquitine/TDP-43 : sclérose latérale amyotrophique

5
Maladie de Huntington

6
Maladie de Huntington Autosomique dominante héréditaire
Expansion anormale des séquences des codons glutamine dans l’ADN (normale 6-35) -- protéine huntingtine Du point de vue clinique : Chorée Démence S’aggrave graduellement et survient plus tôt dans les générations futures d’une famille
 -- protéine huntingtine. Du point de vue clinique : Chorée. Démence. S’aggrave graduellement et survient plus tôt dans les générations futures d’une famille.")
8
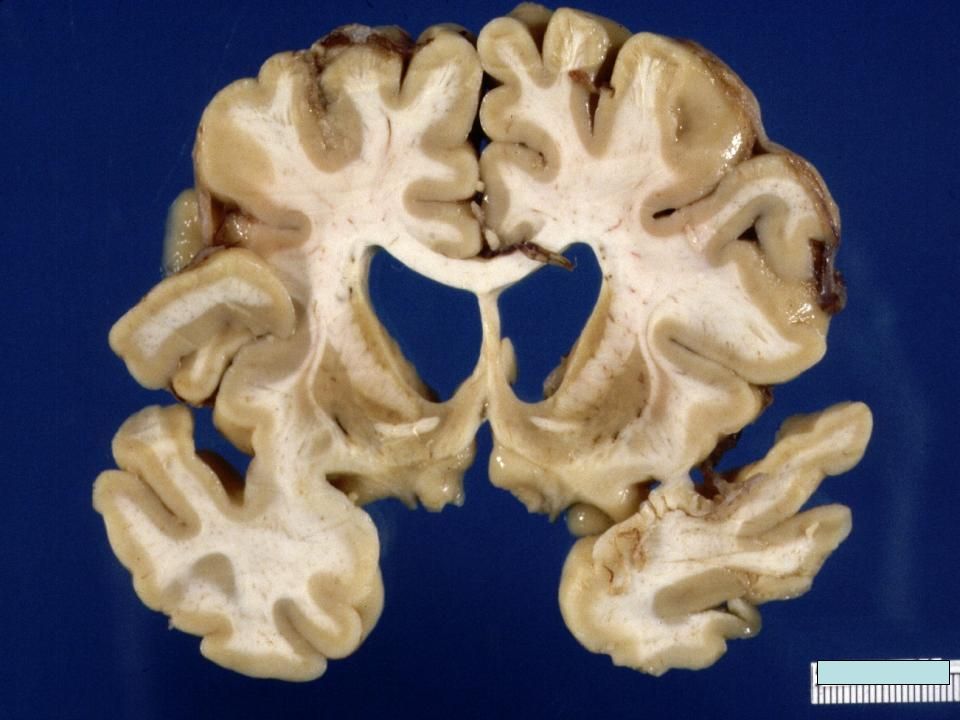
Coupe coronale antérieure
Ventricules élargis (hydrocéphalie ex-vacuo) Noyau caudé avec atrophie sévère Atrophie du putamen
 Noyau caudé avec. atrophie sévère. Atrophie du putamen.")
10
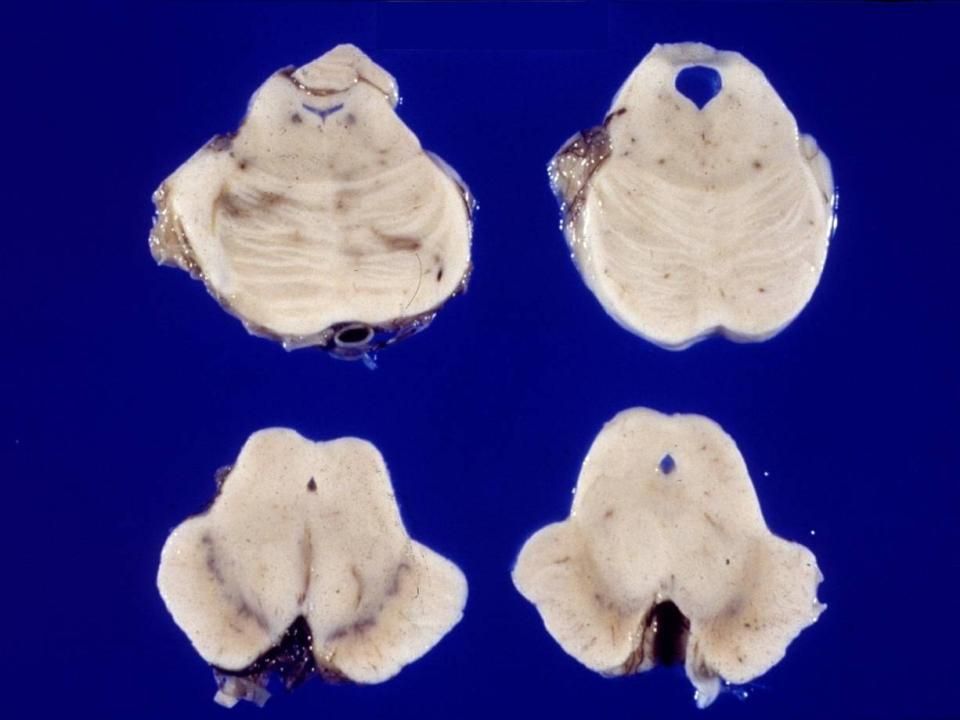
Maladie de Huntington avancée
Putamen normal Maladie de Huntington avancée Fibres striato-pallidales (Wilson’s pencil fibers) Perte des fibres striato-pallidales Perte neuronale et gliosis: ( ne peut être sûr à ce grossissement
 Perte des fibres striato-pallidales. Perte neuronale et gliosis: ( ne peut être sûr à ce. grossissement.")
12
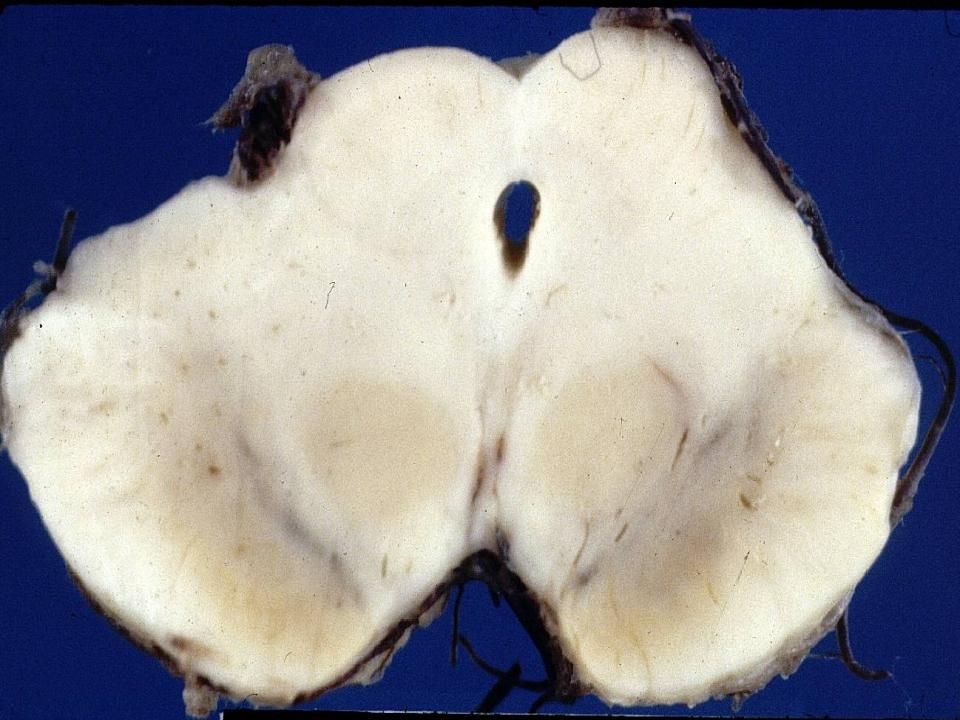
Maladie de Huntington avancée Noyau caudé
Perte neuronale si sévère que le tissu se désintègre avec dégénérescence kystique

13
Maladie de Parkinson

14
Description de cas Homme de 64 ans
Antécédents : Souffrait depuis 6 ans de tremblements progressifs et de rigidité Examen : Élocution lente Changements cognitifs légers (démence) Faciès figé Écoulement de bave Tremblement notable des mains et léger tremblement des jambes au repos Rigidité en roue dentée dans les quatre membres Posture penchée et démarche traînante Est décédé d’une bronchopneumonie à la suite d’une prostatectomie pour un cancer de la prostate
 Faciès figé. Écoulement de bave. Tremblement notable des mains et léger tremblement des jambes au repos. Rigidité en roue dentée dans les quatre membres. Posture penchée et démarche traînante. Est décédé d’une bronchopneumonie à la suite d’une prostatectomie pour un cancer de la prostate.")
16
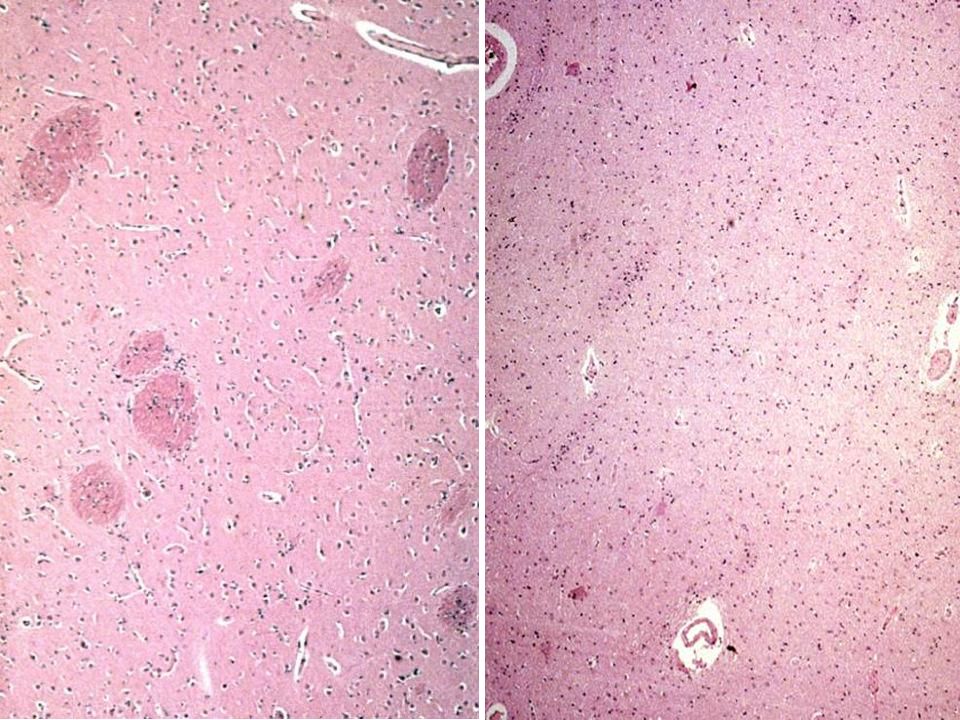
Parkinson Normal Parkinson Normal Locus coeruleus Perte de pigment
donc perte neuronale Locus coeruleus Dilatation de l’aqueduc due à l’atrophie Substantia nigra Parkinson Normal Pigmentation diminuée, SN

17
Mésencéphale H&E α-synucléine

18
Sections du mésencéphale
Sections d’un patient avec maladie de Parkinson dorsal H&E α-synucléine Aqueduc Pédoncule cérébral Substantia nigra Fosse interpédonculaire ventral

19
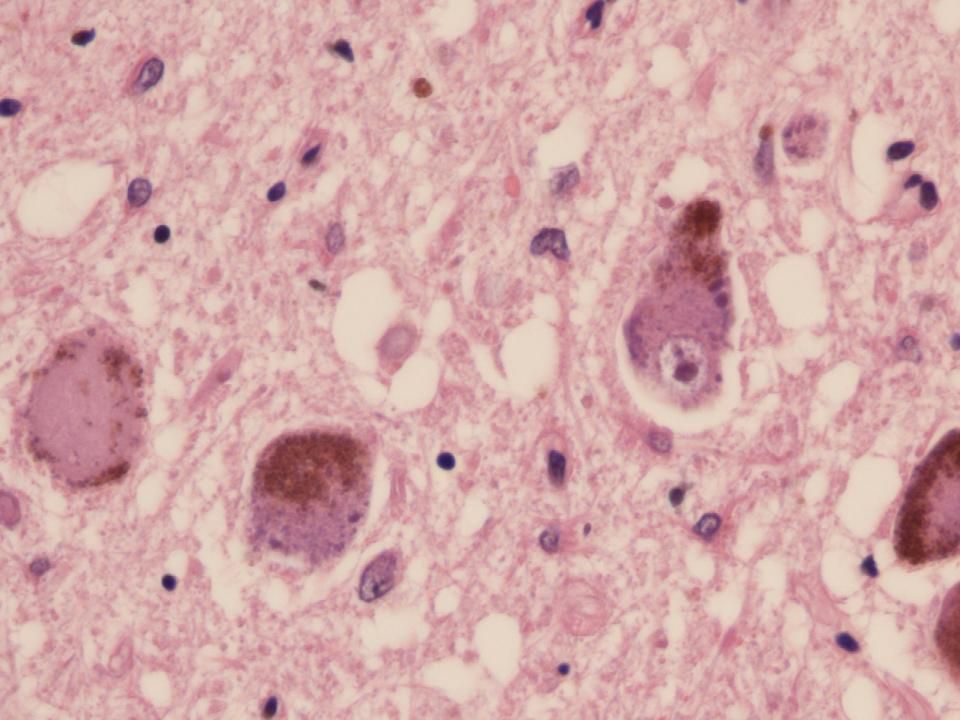
H&E Corps de Lewy

20
Neurones pigmentés SN H&E Corps de Lewy Centre foncé avec halo pâle

21
α-synucléine Corps de Lewy positifs pour l’α-synucléine

22
α-synucléine Corps de Lewy positifs pour l’α-synucléine
Accumulation d’ α-synucléine dans les dendrites

23
Paralysie supranucléaire progressive (PSP)
")
24
PSP Tauopathie—entraîne des enchevêtrements avec dépôts de protéine tau Tauopathie des neurones et des cellules gliales (oligodendrocytes et astrocytes) Maladie neurodégénérative multisystémique Emplacements : Substance grise péri-aqueducale Substance noire Locus ceruleus Noyau sous-thalamique Globus pallidus Substance blanche entourant ces structures
 Maladie neurodégénérative multisystémique. Emplacements : Substance grise péri-aqueducale. Substance noire. Locus ceruleus. Noyau sous-thalamique. Globus pallidus. Substance blanche entourant ces structures.")
25
Signes et symptômes cliniques
Parkinsonisme : Lenteur des mouvements Démarche traînante Tremblement de repos Les médicaments antiparkinsoniens ne sont pas utiles Paralysie du regard vers le bas (entraîne des chutes dans les marches d’escalier) Démence Langage empâté
 Démence. Langage empâté.")
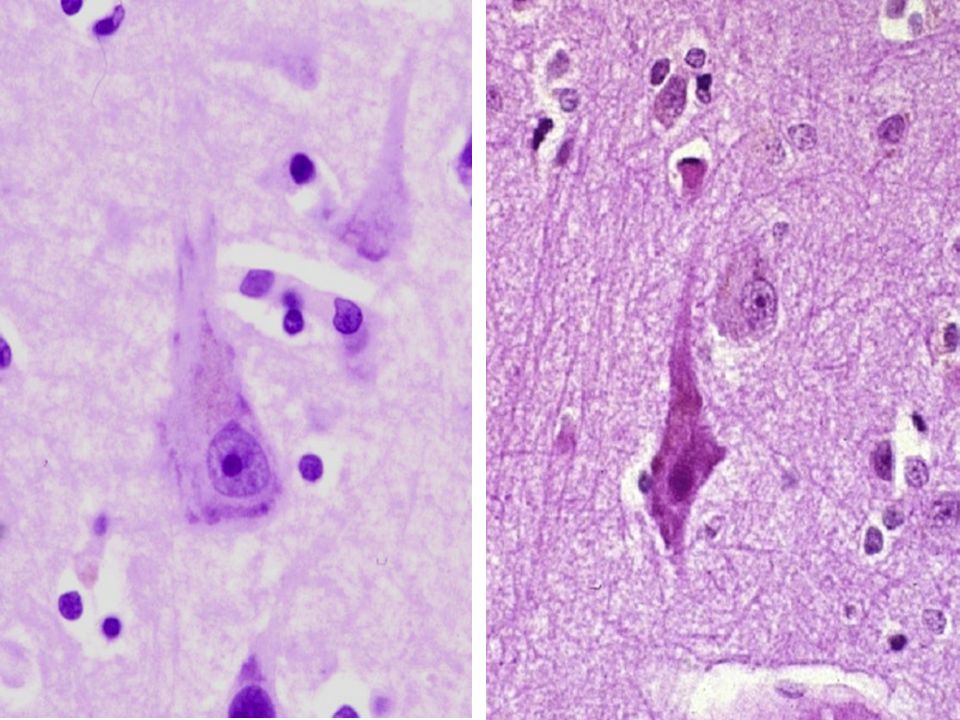
27
PSP Subst. Nigra pâle

29
H&E: Substantia Nigra Neurone pigmenté avec dégénérescence neurofibrillaire: Pigment repoussé en périphérie et aspect en verre dépoli du cytoplasme Neurone pigmenté

31
SN: coloration à l’argent
Neurone gonflé avec dégénérescence neurofibrillaire Neurones pigmentés normaux (cytoplasme non visible avec cette coloration)
")
32
Sclérose latérale amyotrophique

33
Symptômes cliniques/résultats de laboratoire
Affection surtout sporadique, 1 % autosomique dominante Personnes d’âge moyen Faiblesse musculaire dans la partie distale des membres avec fasciculations et atrophie (neurogène) Spasticité (avec signe de Babinski) Incontinence (souvent plus tard) À la fin, insuffisance respiratoire et décès EMG : potentiels géants moteurs et fasciculations (activité spontanée) Sites des anomalies : Cortex moteur (cellules de Betz) Moëlle épinière (tractus pyramidal, neurones des cornes antérieures, racines antérieures ) Muscles (secondaire : atrophie neurogène)
 Spasticité (avec signe de Babinski) Incontinence (souvent plus tard) À la fin, insuffisance respiratoire et décès. EMG : potentiels géants moteurs et fasciculations (activité spontanée) Sites des anomalies : Cortex moteur (cellules de Betz) Moëlle épinière (tractus pyramidal, neurones des cornes antérieures, racines antérieures ) Muscles (secondaire : atrophie neurogène)")
35
Vue supérieure du cerveau
Gyrus post-central (sensitif) Frontal Occipital Gyrus pré-central (moteur) atrophié
 Frontal. Occipital. Gyrus pré-central. (moteur) atrophié.")
37
Neurone moteur normal (cellule de
Betz) du gyrus pré-central Cellule de Betz atrophique dans la SLA
 du gyrus pré-central. Cellule de Betz atrophique. dans la SLA.")
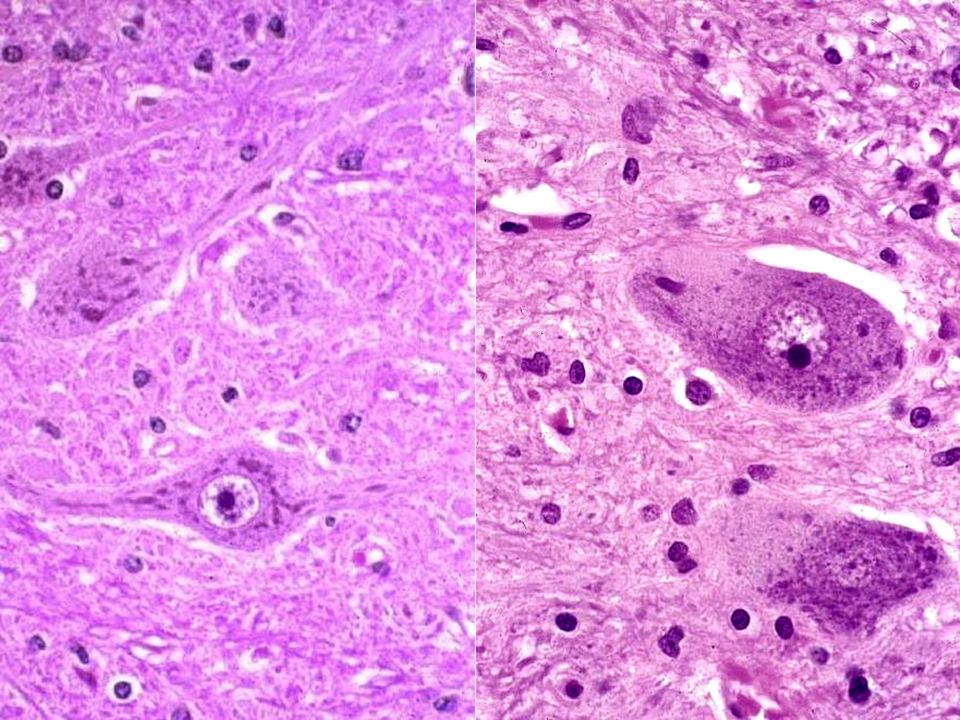
39
SLA: moëlle lombaire, vue antérieure Moëlle normale Cas de SLA Racines nerveuses antérieures plus minces que les dorsales Normalement, même calibre Racine nerveuse dorsale Fx cortico-spinaux latéraux blancs car gliotiques Artère spinale antérieure

41
Neurone moteur, cas de SLA
Neurone moteur normal, corne antérieure, moëlle épinière Corps de Bunina

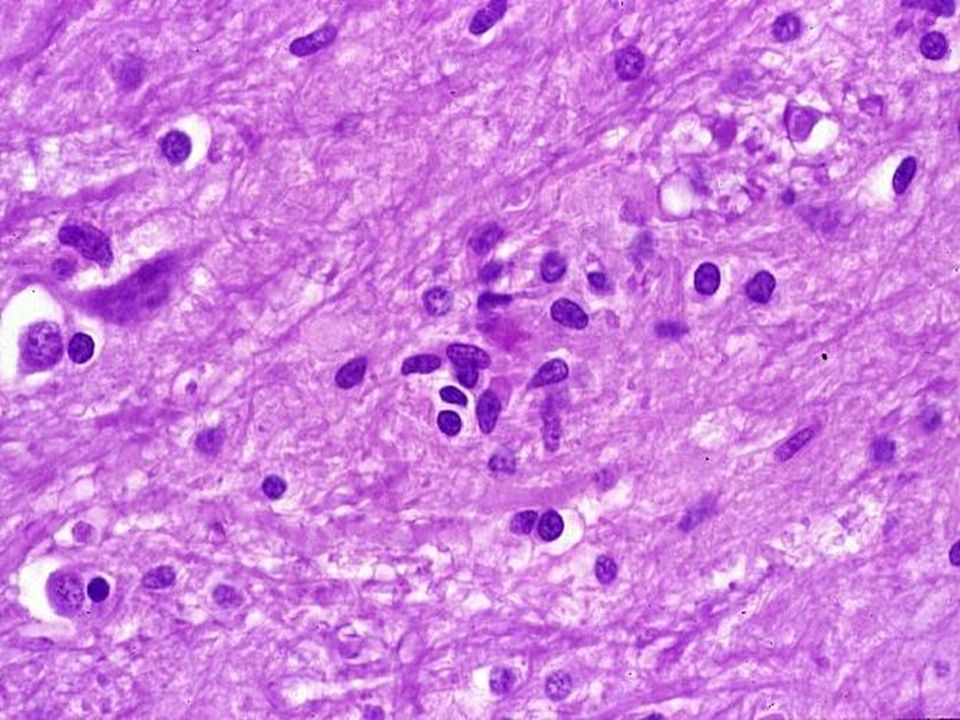
43
Neuronophagie Vestige neuronal Macrophages/cellules microgliales

44
Muscles

45
Maladies musculaires Changements primaires : Changements secondaires :
Inflammation Lésions primaires (dystrophies et myopathies congénitales) Maladies métaboliques (p. ex. glycogénoses, affections mitochondriales) Changements secondaires : Atrophie neurogène (causée par la perte d’innervation et de réinnervation)
 Maladies métaboliques (p. ex. glycogénoses, affections mitochondriales) Changements secondaires : Atrophie neurogène (causée par la perte d’innervation et de réinnervation)")
46
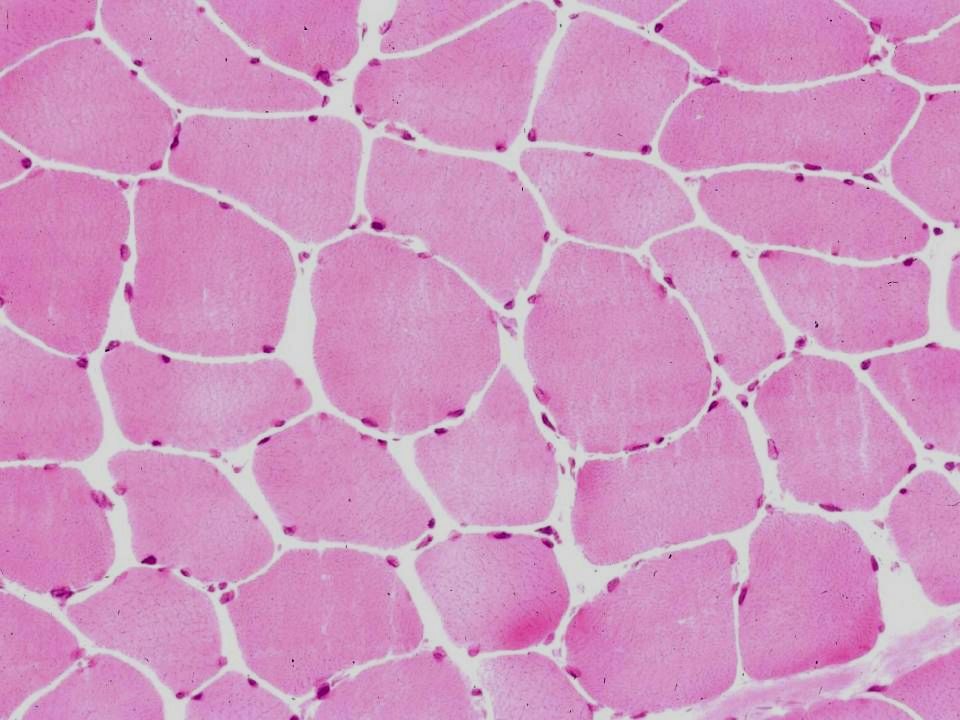
Muscle normal

48
Muscle normal Endomysium Fibres musculaires
Multiples noyaux sous membrane cellulaire

50
Microscopie électronique du muscle (normal)
Bande Z Bande A Bande I Filaments d’actine Filaments de myosine sarcomère

52
NADH H&E Myosine ATP-ase Enzyme oxydatif: fibres de type 1
foncées et de type 2 pâles Myosine ATP-ase Ici pH 9.4: fibres de type 2 foncées et de type 1 pâles H&E

53
Atrophie neurogène

55
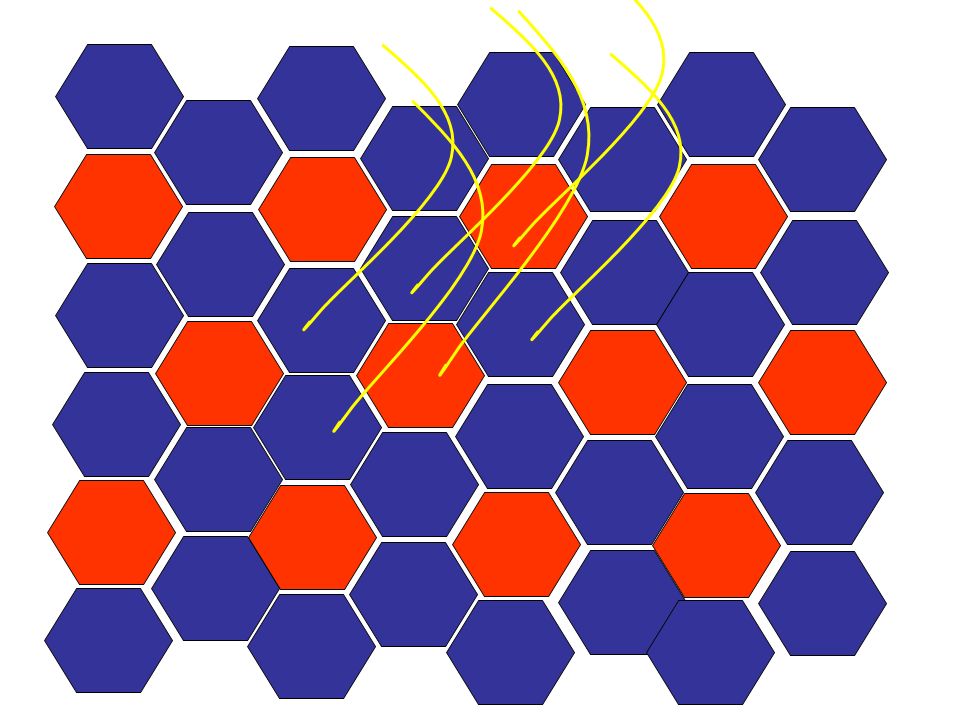
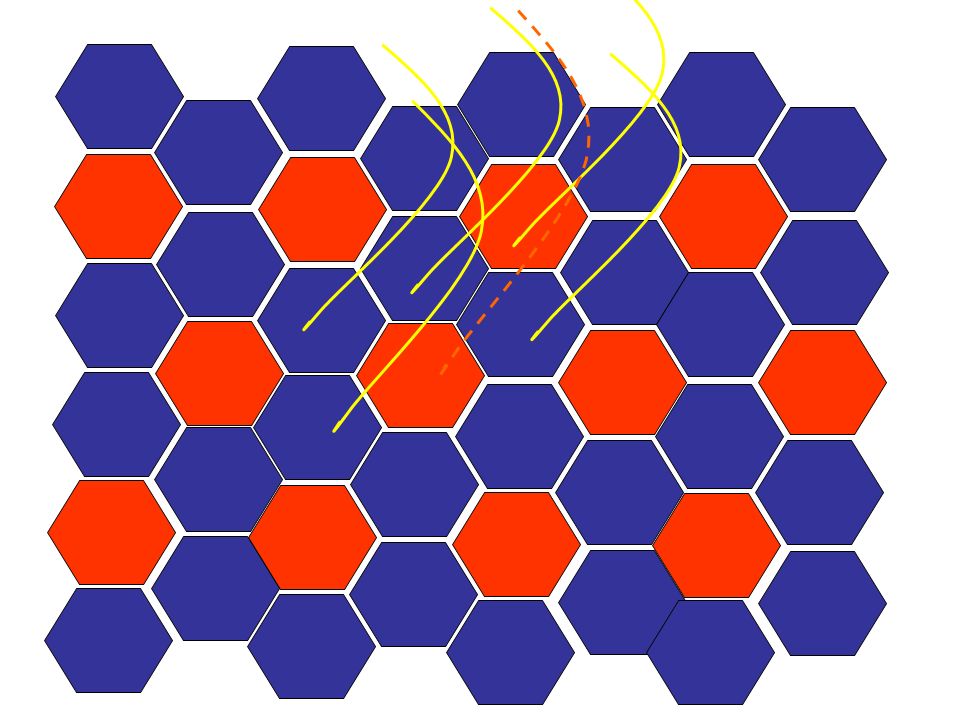
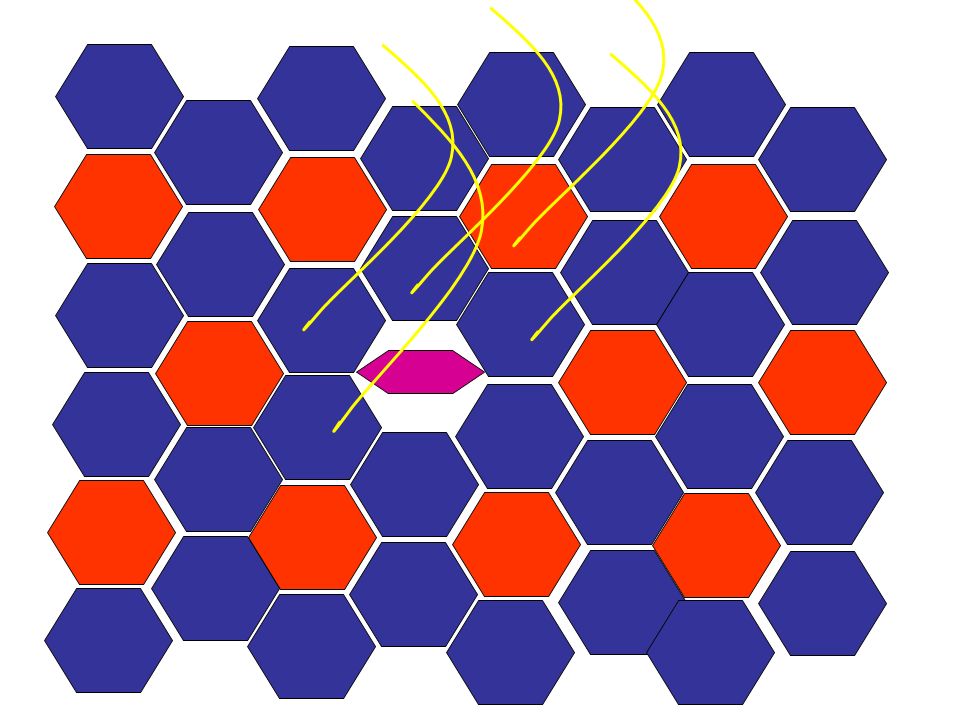
Cette série de diapos est une animation qui vous montre comment l’atrophie neurogène évolue vers les aspects clé d’atrophie de groupe (group atrophy) et de regroupement de type (type grouping). Le muscle normal a un arrangement au hasard de deux types de fibres. L’existence d’un type ou de l’autre est dépendant du type d’innervation (neurone moteur de la corne antérieure latérale = action = fibre de type 2; de la corne antérieure médiane = tonus/position = fibre de type 1) Avec le temps, la distribution initiale des fibres change au fur et à mesure qu’elles perdent leur innervation dans un neuropathie axonale chronique, deviennent atrophiques et reprennent une innervation d’une branche voisine, et ainsi redeviennent de volume normal mais souvent en changeant de type. Avec le temps, l’unité motrice augmente de volume et quand un nouvel axone dégénère, un plus grand groupe de fibres atrophiques est créé (atrophie de groupe) et, ultimement, plus de fibres du même type se retrouvent ensemble (regroupement de type).
 Avec le temps, la distribution initiale des fibres change au fur et à mesure qu’elles perdent leur innervation dans un neuropathie axonale chronique, deviennent atrophiques et reprennent une innervation d’une branche voisine, et ainsi redeviennent de volume normal mais souvent en changeant de type. Avec le temps, l’unité motrice augmente de volume et quand un nouvel axone dégénère, un plus grand groupe de fibres atrophiques est créé (atrophie de groupe) et, ultimement, plus de fibres du même type se retrouvent ensemble (regroupement de type).")
75
H&E d’une atrophie neurogène marquée
Atrophie de groupe Noyaux internes (pas sous la membrane cellulaire): surviennent dans les maladies myopathiques et l’atrophie neurogène sévère Fibres hypertrophiques
: surviennent. dans les maladies myopathiques et. l’atrophie neurogène sévère. Fibres hypertrophiques.")
77
ATP-ase Regroupement de type de fibres Atrophie de groupe

78
Histoire d’un cas Homme de 53 ans
Difficultés pour garder les objets et pour agripper les rampes; échappe les sous Faiblesse des jambes A noté des mouvements musculaires sous la peau (vers) Investigation a montré une atrophie des muscles de la main et une hyper-réflexie.
 Investigation a montré une atrophie des muscles de la main et une hyper-réflexie.")
79
ATPase pH 4.6 H&E

80
ATP-ase pH 4.6 H&E Fibres allongées petites = atrophie de groupe
Perte de l’aspect en échiquier = regroupement des types de fibres Note: ATP-ase avec pH spécial montrant trois types de fibres (à ne pas retenir)
")
82
Images précédentes à plus fort grossissement: mêmes changements.
Le diagnostic de cette biopsie est: atrophie neurogène. Avec les données cliniques, le diagnostic proposé est SLA

84
Dans la SLA et l’atrophie musculaire spinale,
la perte de neurones antérieurs peut être si dramatique que des faisceaux entiers deviennent atrophiques. La réinnervation ne survient pas et ils demeurent ainsi. Faisceaux complets atrophiés

85
Myopathies inflammatoires

86
Myopathies inflammatoires
Dermatomyosite Polymyosite Myosite à corps inclusions

87
Différences DM PM MCI Apparition Précoce Tardive H/F F>H H>F
Faiblesse Proximale Distale Pathologie typique Atrophie périfasciculaire Envahissement des fibres individuelles Vacuoles Sensibilité aux stéroïdes Oui Non

88
Dermatomyosite

89
Dermatomyosite Éléments clé de la dermatomyosite:
Atrophie périfasciculaire Infiltrat inflammatoire mono- nucléé dans le périmysium

90
Polymyosite

91
Polymyosite Éléments clé dans la polymyosite:
Atteinte des fibres au hasard Fibres envahies par des lymphocytes Fibres nécrotiques Pas de vacuoles (trouvées dans la MCI)
")
92
Dystrophies musculaires
Caractéristiques : Nécrose des fibres musculaires individuelles Accumulation de tissu conjonctif autour des fibres musculaires Protéine structurale habituellement manquante (anomalie génétique)
")
93
Dystrophie musculaire de Duchenne (DMD)
")
94
DMD Liée au sexe (hommes atteints, femmes porteuses)
Apparition précoce (vers l’âge de 4 ans) Faiblesse dans les muscles quadriceps et gastrocnémiens d’abord, puis dans tous les muscles proximaux Souvent, la région du muscle gastrocnémien est enflée au stade précoce de la maladie Si non traitée (support respiratoire), le décès survient habituellement vers l’âge de 18 ans
 Faiblesse dans les muscles quadriceps et gastrocnémiens d’abord, puis dans tous les muscles proximaux. Souvent, la région du muscle gastrocnémien est enflée au stade précoce de la maladie. Si non traitée (support respiratoire), le décès survient habituellement vers l’âge de 18 ans.")
96
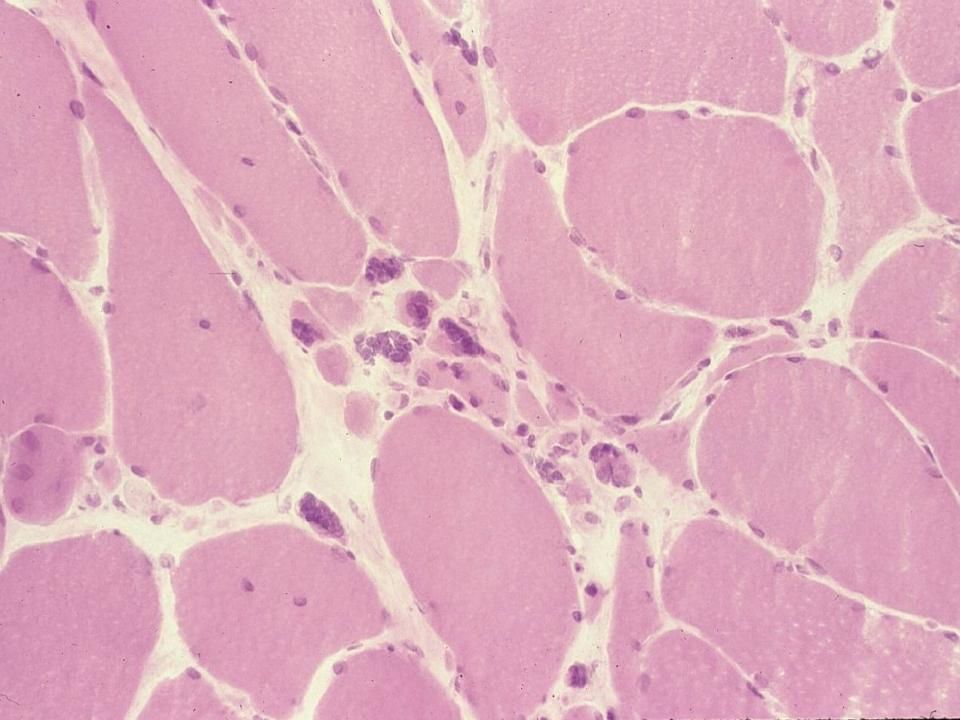
Dystrophie musculaire de Duchenne
Fibre nécrotique envahie par des macrophages Fibre atrophique ronde Fibre nécrotique remplacée par une cellule adipeuse

98
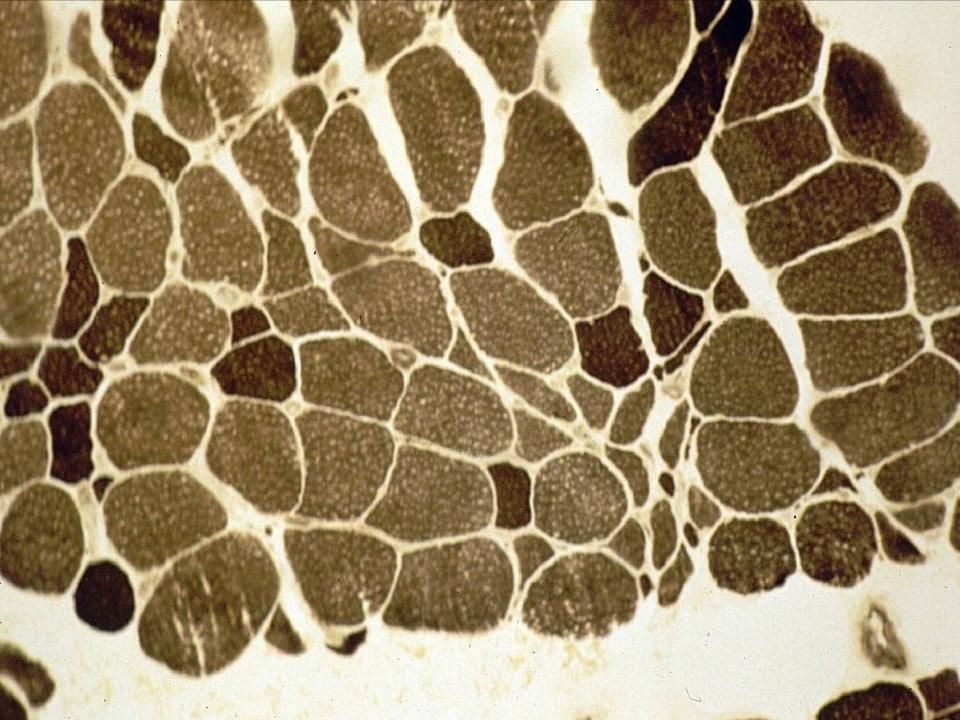
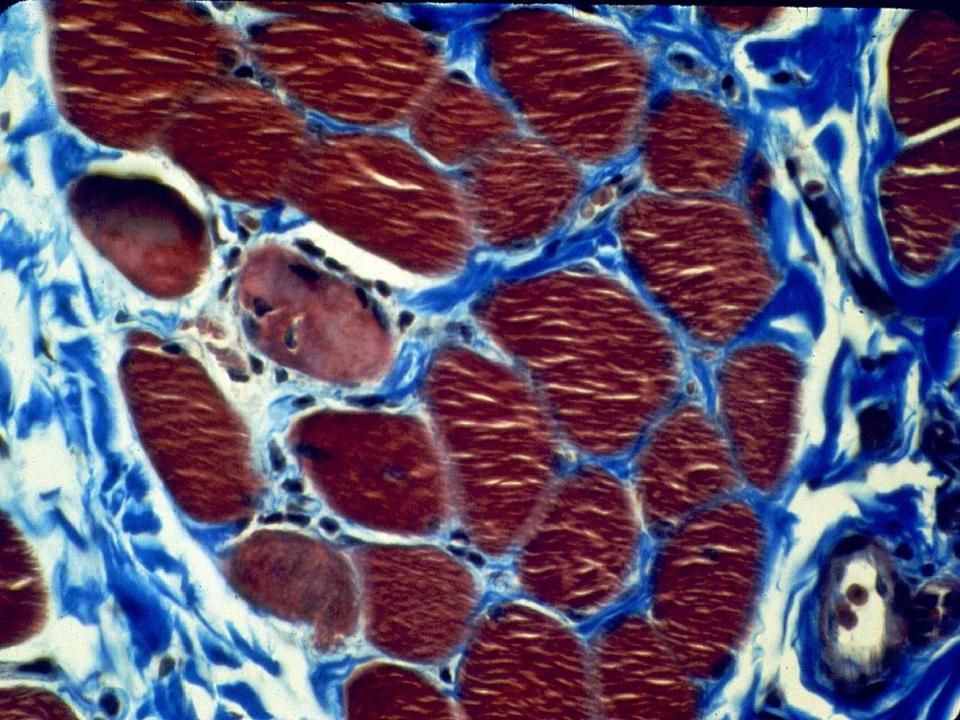
Coloration pour le collagène le montrant en bleu.
Collagène/tissue conjonctif augmenté dans l’endomysium (Duchenne)
")
100
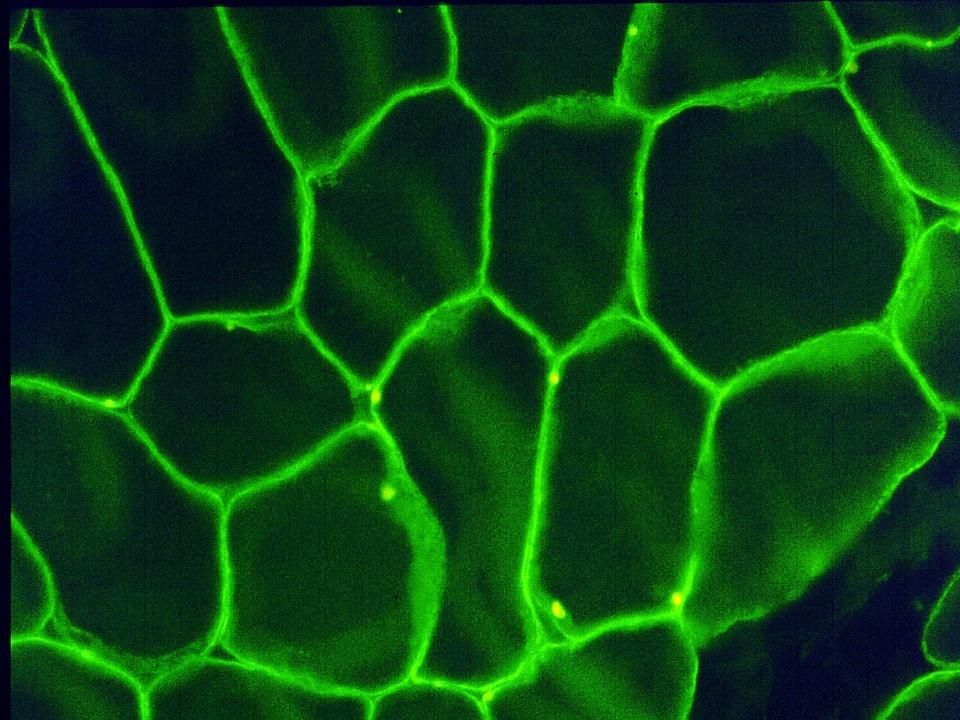
Anticorps anti-dystrophine (fluorescence)
C’est un contrôle normal: la dystrophine est une protéine de la membrane cellulaire qui relie les sarcomères à la membrane cellulaire, évitant une rupture de cette membrane

102
Coloration anti-dystrophine (Duchenne)
La plupart des fibres sont négatives au niveau de la membrane De rare fibres sont positives par l’expression d’utrophine, une forme embryonaire de dystrophine.

Présentations similaires





















>")

>")




>")



>")
 et l’autre sur le muscle en avant de la jambe (le jambier) au cours de mouvements en extension.>")



