Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Aspects Génétiques des Maladies Neuromusculaires
Enseignement complémentaire Maladies neuromusculaires Aspects Génétiques des Maladies Neuromusculaires Modes de transmission et conseil génétique Diagnostic biologique moléculaire Principes de thérapie génique Annabelle Chaussenot Praticien Hospitalier Service de Génétique Médicale Hôpital Archet - CHU de Nice

2
Quelques rappels Deux génomes :
ADN en majorité dans le noyau, fragmentée en chromosomes Petite partie de l’ADN dans les mitochondries NOYAU Mitchondrie

3
Les chromosomes 23 paires dont 22 paires d’autosomes
2 chromosomes sexuels : ♀ 46, XX ♂ 46, XY

4
ADN : Acide Désoxyribonucléique support de l’information génétique
Constitué d’une succession de Nucléotides : A (Adénine) T (Thymine) C (Cytosine) G (Guanine) Appariement complémentaire des nucléotides => formation d’une double hélice à partir des 2 brins
 T (Thymine) C (Cytosine) G (Guanine) Appariement complémentaire des nucléotides. => formation d’une double hélice à partir des 2 brins.")
5
Organisation de l’ADN : condensé sous forme de chromosome

6
Information génétique
Génome : ensemble du matériel génétique d'un individu ou d'une espèce codé dans son ADN => 3 milliards de paires de bases

7
Les gènes Succession d’intron et d’exon

8
Les mutations X gène gène messager messager Protéine Protéine
Définition : Variation de la séquence ADN sur un gène entraînant une absence de production ou une modification de la protéine qui sera responsable de la maladie Plusieurs types de mutation: Ponctuelle: variation sur un nucléotide => faux-sens ; non-sens Délétion : variation sur un ou plusieurs nucléotides voir plusieurs exons en phase ou non => décalage du cadre de lecture amenant à un codon stop prématuré

9
Différents modes de transmission
Père Mère Tous les chromosomes sont regroupés par paires => 2 allèles d’un gène Chaque parent transmet une copie à son enfant. Il existe 4 possibilités de combinaison pour une paire de chromosomes et donc pour les 2 allèles d’un gène chez les enfants. les enfants

10
Maladies de transmission Autosomiques Dominantes
Exemples : Maladie de Steinert Maladie de Charcot-Marie IA Dystrophie Facio-Scapulo-Humérale Dominant : caractère qui s’exprime dès qu’il est présent sur une des 2 copies du gène (hétérozygote).
.")
11
Autosomique Dominant Père Mère
Lorsque la maladie est autosomique dominante, un seul gène non fonctionnel suffit pour que la maladie s’exprime. Un sujet atteint a un risque de 50% de transmettre la maladie à chaque enfant Autant d’hommes que de femmes 50% malades 50% sains

12
Quelques nuances Pénétrance incomplète Expressivité variable
Phénomène d’anticipation Mutation De Novo Mosaïcisme germinale

13
Pénétrance Individus atteints Complète : Tous les patients porteurs de la mutation développeront la maladie Incomplète : Certains individus porteurs de la mutation ne développent pas la maladie possibilité de « sauts de génération ». Individus porteurs asymptomatiques Pénétrance = 6/12 = 50 %

14
Expressivité variable
Degré de sévérité de la maladie variable Tous les signes cliniques ne sont pas systématiquement présents Expression de la maladie varie selon le sexe du parent atteint: Transmission maternelle des formes néonatales dans la dystrophie myotonique de Steinert : Début âge adulte : calvitie, cataracte => myopathie, myotonie, troubles cardiaques, hypogonadisme Forme congénitale : forme très grave d’expression néonatale

15
Mutation instable avec phénomène d’anticipation
La gravité de la maladie est fonction du nombre de répétition (CTG)n Ex :Maladie de Steinert Normaux : 5-40, prémutés : 50-80, mutés : >80 55 asymptomatique Calvitie, cataracte,… 65 60 Adulte : Myotonie, trouble de la conduction,... Enfant: myopathie + déficit intellectuel 85 400 480 Forme néonatale grave avec décès précoce 500 2000 1100 1200
n. Ex :Maladie de Steinert. Normaux : 5-40, prémutés : 50-80, mutés : > asymptomatique. Calvitie, cataracte,… Adulte : Myotonie, trouble de la conduction,... Enfant: myopathie + déficit intellectuel Forme néonatale grave avec décès précoce")
16
La maladie de Steinert ou dystrophie myotonique de type 1
La plus fréquente des dystrophies musculaires de l’adulte (Prévalence : 1/8 000) La forme adulte débute entre 30-40ans et est caractérisée par : Myotonie : lenteur anormale et indolore à la décontraction musculaire ++ mains Déficit musculaire : faciès myopathique (ptosis, sourire transversale) Troubles du rythme et/ou de conduction cardiaque (risque de mort subite ++) Cataracte, Atteinte endocrinienne (diabète, hypogonadisme, azoospermie) Troubles neurologiques centraux (hypersomnie, troubles cognitifs) Calvitie précoce. Evolution lentement progressive mais une dégradation rapide peut parfois être observée. Mortalité accrue par complications pulmonaires et cardiaques Génétique: Transmission autosomique dominante avec un phénomène d’anticipation. Conseil génétique ++délicat : grande variabilité d’expression clinique inter- et intrafamiliale. Transmission maternelle des formes très grave néonatales : ++ diagnostic prénatal
 La forme adulte débute entre 30-40ans et est caractérisée par : Myotonie : lenteur anormale et indolore à la décontraction musculaire ++ mains. Déficit musculaire : faciès myopathique (ptosis, sourire transversale) Troubles du rythme et/ou de conduction cardiaque (risque de mort subite ++) Cataracte, Atteinte endocrinienne (diabète, hypogonadisme, azoospermie) Troubles neurologiques centraux (hypersomnie, troubles cognitifs) Calvitie précoce. Evolution lentement progressive mais une dégradation rapide peut parfois être observée. Mortalité accrue par complications pulmonaires et cardiaques. Génétique: Transmission autosomique dominante avec un phénomène d’anticipation. Conseil génétique ++délicat : grande variabilité d’expression clinique inter- et intrafamiliale. Transmission maternelle des formes très grave néonatales : ++ diagnostic prénatal.")
18
Maladie de Steinert La gravité de la maladie est fonction du nombre de répétition
55 65 4 formes cliniques qui diffèrent par l’âge de début et la sévérité statistiquement corrélées au nombre de répétitions de triplets CTG. Mais de nombreux cas échappent à la “règle” ! 60 85 400 480 500 2000 1100 1200 asymptomatique Forme bénigne : le plus souvent entre 50 et 150 Calvitie, cataracte,… Forme Adulte : Myotonie, trouble de la conduction,... => entre 300 et 1000 Forme juvénile: atteinte musculaire plus discrète / ++ atteinte cognitive et comportementale => en général entre Forme néonatale grave avec décès précoce : arthrogryppose, retard mental, décès précoce => > 1500

19
Mutations de novo +/+ +/+ +/+ +/+ +/+ +/+ +/+ +/+ +/+ m/+ +/+ +/+ +/+

20
Mosaïques germinales Mosaïque : présence de deux populations de cellules, l’une étant porteuse d’une mutation, l’autre non +/+ +/+ +/+ +/+ +/+ +/+ Individu portant une mutation à l’état mosaïque +/+ m/+ +/+ m/+ +/+ m/+ m/+ +/+ ++ Myopathie de Duchenne

21
Maladie de transmission Autosomique Récessive
Récessif : caractère qui s’exprime seulement lorsqu’il est présent sur les 2 copies du gène (homozygote) Exemples : Amyotrophie spinale infantile Ataxie de Friedreich Maladies métaboliques
 Exemples : Amyotrophie spinale infantile. Ataxie de Friedreich. Maladies métaboliques.")
22
50% de porteurs «sains» hétérozygotes
Autosomique Récessif Père Mère Lorsque la maladie est autosomique récessive, il faut que les 2 chromosomes transmis portent un même gène non fonctionnel pour que la maladie s’exprime. Gène non fonctionnel Enfants Parents indemnes et hétérozygotes, le risque est de 25% de transmettre la maladie à chaque enfant (touche 1 fratrie) Touche également les deux sexes 50% de porteurs «sains» hétérozygotes 25% malades 25% sains
 Touche également les deux sexes. 50% de porteurs «sains» hétérozygotes. 25% malades. 25% sains.")
23
Maladie de transmission Autosomique Récessive
Homozygotes : les 2 copies du gène sont porteurs d’une mutation recessive et la maladie s’exprime Hétérozygotes : 1 copie du gène porte une mutation recessive, l’autre copie est normale

24
Risque de transmission de la maladie pour un couple et pas pour un individu seul
Risque pour les parents d’un enfant atteint : 1 / 4 Risque pour les couples apparentés est fonction de : Fréquence de la maladie : détermine la fréquence des hétérozygotes dans la population générale Et donc le probabilité pour que le conjoint soit hétérozygote Fréquence de la maladie ou de certaines mutations en fonction des origines géographiques Consanguinité : union de sujets apparentés (plus de risque d’être porteurs du même allèle récessif originaire de l’ancêtre commun)
")
25
Risque varie en fonction de la fréquence de la maladie
Ensemble des maladies rares Très rares Fréquentes Amyotrophie spinale infantile : Fréquence de la maladie : 1/6000 Fréquence des hétérozygote : 1/40 1 personne sur 40 dans la population générale est hétérozygote Myopathie de Nonaka : Fréquence de la maladie : 1/ Fréquence des hétérozygote : 1/500 1 personne sur 500 dans la population générale est hétérozygote

26
Exemple 1 : ASI ½ ¼ 2/3 x 1/40 x 1/4 = 1/240 Risque d’être atteint
= 1/320 2/3 x 1/40 x 1/4 = 1/240

27
Exemple 2 => FKRP alpha-dystroglycanopathie
Risque d’être hétérozygote dans la population générale : 1/120 => FKRP alpha-dystroglycanopathie

28
Transmission Récessive liée à l’X
Caractère déterminé par des gènes situé sur le chromosome X Femmes conductrices, exceptionnellement symptomatiques (homozygotie, syndrome de turner, X inactivée) Exemples: Myopathies de Duchenne et Becker Myopathie d’Emery-Dreyfuss
 Exemples: Myopathies de Duchenne et Becker Myopathie d’Emery-Dreyfuss.")
29
25% filles conductrices non malade
Lié à l’X Père Mère Les chromosomes sexuels X et Y déterminent le sexe de l’enfant. La maladie s’exprime seulement chez les garçons. Les femmes sont conductrices, c’est-à-dire qu’elles transmettent le chromosome X porteur du gène non fonctionnel. XY XX XX XX XY XY 25% filles saines 25% filles conductrices non malade 25% garçons sains 25% garçons malades

30
Transmission Dominante liée à l’X
Trait porté par des gènes du chromosome X qui se manifeste chez les hommes et chez les femmes Très rare : Charcot-Marie-Tooth dominante liée à l’X Déficit en Ornithine transcarbamylase

31
Transmission Maternelle liée à l’ADN Mitochondrial
Homme et femme en général atteint de la même manière Transmis uniquement par les femmes Aucune transmission père - enfant

32
Les maladies mitochondriales : =anomalie de la chaine respiratoire
Chromosomes ou ADN nucléaire La Cellule La Mitochondrie ADN mitochondrial Membrane mitochondriale La Chaîne Respiratoire mitochondriale GLUCIDE + LIPIDES III IV V I II ENERGIE Les maladies mitochondriales : =anomalie de la chaine respiratoire ≠ ADN mitochondrial

33
ADNmt ADN nucléaire Assemblage C I C II C III C IV C V
Protéines d’origine mitochondriale Assemblage C I C II C III C IV C V Protéines d’origine nucléaire

34
Pathologies mitochondriales liées à l’ADN mitochondrial 2 particularités
Transmission uniquement maternelle Hétéroplasmie : Présence dans une même cellule des copies d'ADNmt normales et mutées => proportion d’ADN muté ++variable d'une cellule à l'autre, d'un organe à l'autre, et au cours du temps => hétérogénéité clinique Pourquoi ? Plusieurs mitochondries ( ) par cellule Plusieurs copies d'ADNmt (2 à 10) par mitochondrie Pourquoi ? Plusieurs dizaines à milliers de mitochondries dans chaque cellule Chaque mitochondrie possède plusieurs copies d'ADNmt (2 à 10) Au cours des divisions cellulaires, leur distribution se fait au hasard => grande variation de leur proportion d'une cellule à l'autre, d'un organe à l'autre, et au cours du temps Distribution se fait au hasard lors des divisions cellulaires Distribution se fait au hasard lors des divisions cellulaires
 par cellule. Plusieurs copies d ADNmt (2 à 10) par mitochondrie. Pourquoi Plusieurs dizaines à milliers de mitochondries dans chaque cellule. Chaque mitochondrie possède plusieurs copies d ADNmt (2 à 10) Au cours des divisions cellulaires, leur distribution se fait au hasard. => grande variation de leur proportion. d une cellule à l autre, d un organe à l autre, et au cours du temps. Distribution se fait au hasard lors des divisions cellulaires. Distribution se fait au hasard lors des divisions cellulaires.")
35
Analyses génétiques Intérêts et Limites

36
Intérêts des analyses génétiques
1- Mettre enfin un nom sur sa maladie 2- Bénéficier d’une prise en charge optimale et adaptée 3- Accéder à un conseil génétique fiable 4- Être éventuellement inclus dans des essais thérapeutiques 5- Pour faire progresser les connaissances sur la maladie

37
Révision et Précision du diagnostic
Pourquoi un diagnostic peut-il être erroné ou imprécis ? - Similitude clinique avec une autre maladie / Atypie de la symptomatologie - Evolution clinique avec le temps - Absence à l'époque de technologies modernes d'analyse - Indisponibilité des tests génétiques à l'époque - Gène lié à la maladie non encore identifié du diagnostic - Gène identifié mais difficile à étudier (très grand et/ou très complexe) Le diagnostic moléculaire des maladies génétiques: Objectif: établir un diagnostic précis => par l’identification de l’anomalie génétique Intérêt: certitude diagnostique => prise en charge adaptée => conseil génétique
 Le diagnostic moléculaire des maladies génétiques: Objectif: établir un diagnostic précis. => par l’identification de l’anomalie génétique. Intérêt: certitude diagnostique. => prise en charge adaptée. => conseil génétique.")
38
Accéder à un conseil génétique fiable
Conseil génétique: INFORMER du risque de transmettre la maladie à sa descendance, ou du risque d’avoir un autre enfant atteint mais également le risque pour les autres membres de la famille Nouveau diagnostic = Conseil génétique différent pour toute la famille Que peut-on proposer quand il existe un diagnostic génétique précis ? Diagnostic prénatal ou préimplantatoire Diagnostic symptomatique rapide Diagnostic présymptomatique

39
Éléments nécessaires au conseil génétique
Mode de transmission Autosomique dominant Autosomique récessif Récessif lié à l’X Dominant lié à l’X Mitochondrial Quelques nuances Pénétrance : complète / incomplète ? Expressivité : variable ? Phénomène d’anticipation ? Mutation De Novo Mosaïcisme germinale => Conseil génétique spécifique pour chaque maladie génétique, voir pour chaque famille

40
Les difficultés et les limites de la génétique

41
Tests génétiques Laboratoire de biologie moléculaire (réseau en France) Tous ne réalisent pas tous les tests génétiques, pour toutes les maladies ! => analyses complexes et longues => laboratoires spécialisés => dans un cadre de recherche => délais très longs Du plus simple au plus compliqué Soit les tests génétiques sont facilement accessibles, et une simple prise de sang suffit ! Amyotrophie spinale infantile, Maladie de Steinert, myopathie FSH Soit un nouveau bilan complet est nécessaire pour orienter le test génétique : +/- EMG, dosages sanguins, imagerie et biopsie musculaire

42
Les répercussions Les limites
- Anxiété (attente des résultats et peur de l’annonce) - Déception (reste des incertitudes, pas toutes les réponses, pas de traitement curatif) Conseil génétique (conséquence pour les enfants voir les petits-enfants) Les limites Jusqu’où aller dans la précision ? Ex : Amyotrophie spinale proximale Précision: délétion du gène SMN1 => Degré de gravité ? nombre de copies du gène SMN2 Informer les apparentés à risque de transmettre la maladie ou d’être atteint : C’est au malade que revient la responsabilité d’informer les membres de sa famille Des lacunes encore à combler ! Ex : Dystrophies musculaires des ceintures: 22 formes différentes décrites à ce jour 16 gènes identifiés mais encore inconnus pour 6 formes Ex : Myopathie congénitale à bâtonnets (biopsie musculaire chez un nouveau-né hypotonique) 6 gènes responsables connus actuellement (ACTA1, TPM2, TPM3, NEB, CFL2, TNNT1) Travail très long et aléatoire; autres gènes encore non identifiés
 - Déception (reste des incertitudes, pas toutes les réponses, pas de traitement curatif) - Conseil génétique (conséquence pour les enfants voir les petits-enfants) Les limites. Jusqu’où aller dans la précision Ex : Amyotrophie spinale proximale. Précision: délétion du gène SMN1 => Degré de gravité nombre de copies du gène SMN2. Informer les apparentés à risque de transmettre la maladie ou d’être atteint : C’est au malade que revient la responsabilité d’informer les membres de sa famille. Des lacunes encore à combler ! Ex : Dystrophies musculaires des ceintures: 22 formes différentes décrites à ce jour. 16 gènes identifiés mais encore inconnus pour 6 formes. Ex : Myopathie congénitale à bâtonnets (biopsie musculaire chez un nouveau-né hypotonique) 6 gènes responsables connus actuellement (ACTA1, TPM2, TPM3, NEB, CFL2, TNNT1) Travail très long et aléatoire; autres gènes encore non identifiés.")
43
Diagnostic d’une maladie neuromusculaire Un processus parfois long et complexe…
Symptômes Interrogatoire, examen clinique, examens complémentaires (CPK, IRM, EMG…) Un ou plusieurs diagnostics évoqués Tests génétiques Biopsie musculaire Un ou plusieurs diagnostics évoqués Confrontation anatamo- clinique Diagnostic confirmé Diagnostic non confirmé Tests génétiques Négatifs = d’autres gènes sont en cause
 Un ou plusieurs diagnostics. évoqués. Tests génétiques. Biopsie musculaire. Un ou plusieurs diagnostics évoqués. Confrontation anatamo- clinique. Diagnostic. confirmé Diagnostic. non confirmé. Tests génétiques. Négatifs = d’autres gènes sont en cause.")
44
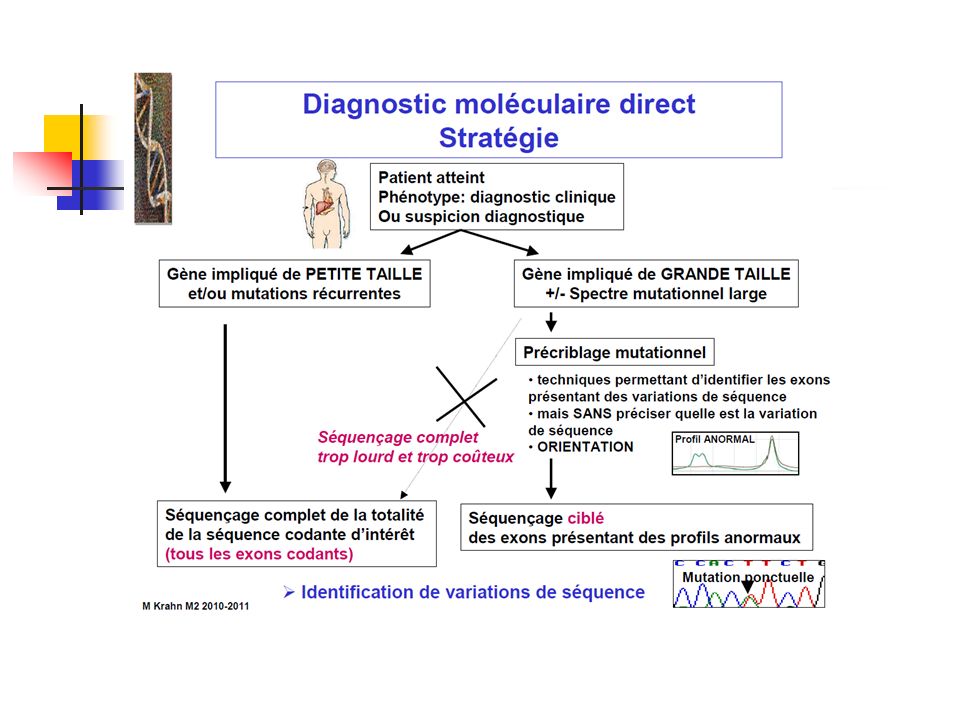
Stratégie générale d’une analyse génétique

46
Nombreuses techniques d’analyses moléculaires
Séquençage du gène RT-PCR Analyse de liaison => recherche de région homozygote Exome Séquençage haut débit avec puces de recaptures

47
Quelques exemples

49
Maladie du motoneurone
Amyotrophies spinales (NMp) Amyotrophie spinale proximale: Récessif => gène SMN1 (SMN2 = modulateur) Maladie de Kennedy: amyotrophie spino-bulbaire progressive +tr endocriniens => gène du Récepteur aux androgènes (lié à l’X) Neuropathies motrices distales héréditaires (AD) HSPB8 , HSPB1, BSCL2, Senataxine, GARS Paraparésies spastiques héréditaires (NMc) Autosomique dominante : SPG4 (40 %) > SPG3A (<20 ans) > SPG31 > SPG10 Autosomique récessive : SPG7 (5 %) > SPG5 , SPG11 et SPG15 Récessive liée à l’X : SPG1 Sclérose Latéral Amyotrophique : 5–10% => +++ AD (parfois AR ou lié à l’X) familial SOD1 : 20% des familles SLA-AD TARDBP (TDP-43) : 2 à 5% C9ORF72 Plus rare : Alsin, Senataxin,VAPB, DCTN1, Angiogenine, MAPT, FUS
 Amyotrophie spinale proximale: Récessif => gène SMN1 (SMN2 = modulateur) Maladie de Kennedy: amyotrophie spino-bulbaire progressive +tr endocriniens. => gène du Récepteur aux androgènes (lié à l’X) Neuropathies motrices distales héréditaires (AD) HSPB8 , HSPB1, BSCL2, Senataxine, GARS. Paraparésies spastiques héréditaires (NMc) Autosomique dominante : SPG4 (40 %) > SPG3A (<20 ans) > SPG31 > SPG10. Autosomique récessive : SPG7 (5 %) > SPG5 , SPG11 et SPG15. Récessive liée à l’X : SPG1. Sclérose Latéral Amyotrophique : 5–10% => +++ AD (parfois AR ou lié à l’X) familial SOD1 : 20% des familles SLA-AD. TARDBP (TDP-43) : 2 à 5% C9ORF72. Plus rare : Alsin, Senataxin,VAPB, DCTN1, Angiogenine, MAPT, FUS.")
50
Maladie de Charcot-Marie-Tooth (CMT) Neuropathies Héréditaires
Démyélinisant => CMT1 VCNm (médian) < 38 m/s Axonal => CMT2 VCNm (médian) > 38 m/s DOMINANT PMP22 P0 LITAF NEFL MFN2 En fonction de certains signes: RAB7, SPTLC1, GARS, LMNA Dominant lié à l’X CX32 RECESSIF GDAP1 KIAA1985 MTMR2 PRX P0, PMP22, EGR2, MTMR13 LMNA CMT1A, … CMT2A, …
 < 38 m/s. Axonal => CMT2. VCNm (médian) > 38 m/s. DOMINANT. PMP22. P0. LITAF. NEFL. MFN2. En fonction de certains signes: RAB7, SPTLC1, GARS, LMNA. Dominant lié à l’X. CX32. RECESSIF. GDAP1. KIAA1985. MTMR2. PRX. P0, PMP22, EGR2, MTMR13. LMNA. CMT1A, … CMT2A, …")
51
SYNDROMES MYASTHENIQUES CONGENITAUX
EMG: Incrément / Dédoublement / Décrément Récessif ColQ RAPSN MUSK CHAT CHRNE Dominant / Récessif CHRNA1 Dominant / Récessif CHRNB1 CHRND SCN4A

52
Canalopathies Paralysies Périodiques Primitives
Syndrome d’Andersen KCNJ2 Paralysie Périodique Hypokaliémique (HOKPP) CACNA1S SCN4A Paralysie Périodique Normo/Hyperkaliémique (HYPP) Myotonies Non Dystrophiques Myotonie congénitale (MC) de Becker ou Thomsen CLCN1 Paramyotonie congénitale (PC) de Von Eulenburg SCN4A Autres myotonies non dystrophiques, dont : - myotonia fluctuans - myotonia permanens
 CACNA1S. SCN4A. Paralysie Périodique Normo/Hyperkaliémique (HYPP) Myotonies Non Dystrophiques. Myotonie congénitale (MC) de Becker ou Thomsen. CLCN1. Paramyotonie congénitale (PC) de Von Eulenburg. SCN4A. Autres myotonies non dystrophiques, dont : - myotonia fluctuans. - myotonia permanens.")
53
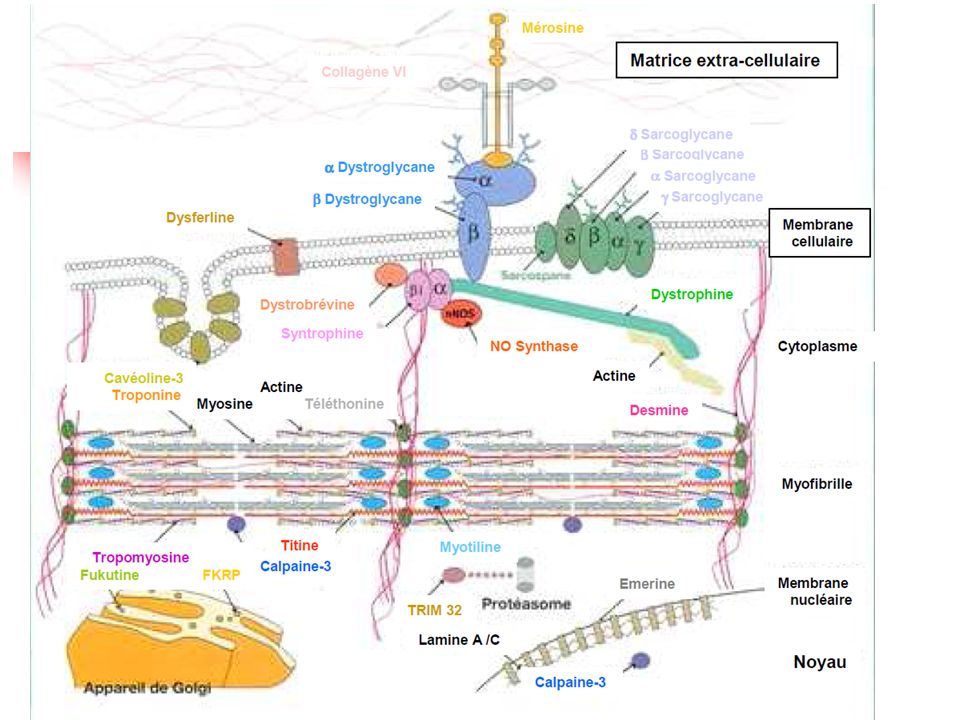
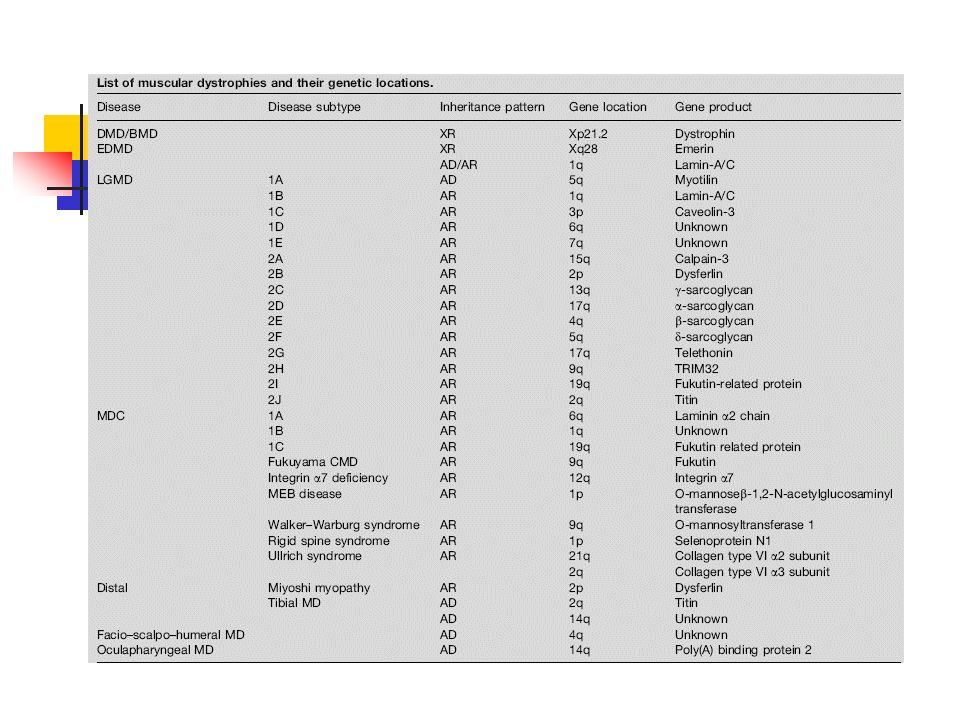
Myologie Dystrophies musculaires Syndromes myotoniques
Dystrophies musculaires de Duchenne / de Becker (BMD) Dystrophies musculaires congénitales Dystrophies musculaires des ceintures (LGMD) Alpha –Sarcoglycanopathies (LGMD2D) Gamma –Sarcoglycanopathies (LGMD2C) Calpainopathies (LGMD2A, Myosites à éosinophiles idiopathiques) Dysferlinopathies (LGMD2B, Myopathie de Miyoshi) Myopathies facio-scapulo-humérales de type Landouzy-Déjerine Syndromes myotoniques Dystrophie myotonique de type 1 (Maladie de STEINERT) / de type 2 (PROMM) Myotonies congénitales non dystrophiques Myopathies congénitales Myopathie centronucléaire Myopathie myotubulaire Myopathie à bâtonnets Myopathie central-core
 Dystrophies musculaires congénitales. Dystrophies musculaires des ceintures (LGMD) Alpha –Sarcoglycanopathies (LGMD2D) Gamma –Sarcoglycanopathies (LGMD2C) Calpainopathies (LGMD2A, Myosites à éosinophiles idiopathiques) Dysferlinopathies (LGMD2B, Myopathie de Miyoshi) Myopathies facio-scapulo-humérales de type Landouzy-Déjerine. Syndromes myotoniques. Dystrophie myotonique de type 1 (Maladie de STEINERT) / de type 2 (PROMM) Myotonies congénitales non dystrophiques. Myopathies congénitales. Myopathie centronucléaire. Myopathie myotubulaire. Myopathie à bâtonnets. Myopathie central-core.")
57
Thérapie génique Stratégie thérapeutique qui consiste à faire pénétrer des gènes dans les cellules ou les tissus d'un individu pour traiter une maladie. => vise à remplacer ou complémenter un allèle mutant défectif par un allèle fonctionnel ou à surexprimer une protéine dont l'activité aurait un impact thérapeutique Vecteurs viraux Vecteurs non-viraux

59
DYSTROPHINE

61
Le saut d’exon X 48 49 50 51 52 48 49 50 51 52 Messager normal
Dystrophine fonctionnelle Délétion de plusieurs exons avec décalage du cadre de lecture amenant à un codon stop prématuré 48 X 49 52 48 49 48 49 52 Pas de dystrophine ou Protéine tronquée Dystrophine légèrement raccourcie mais fonctionnelle

62
Conclusions Importance du diagnostic génétique:
Pour soi-même Pour le conseil génétique de la famille Pour permettre des progrès scientifique en terme de connaissance et de compréhension des maladies, indispensable à la mise au point de traitement. Mais il reste encore de nombreuses choses à découvrir : Des gènes qui n’ont pas encore été identifiés Les mécanismes physiopathologiques L’identification de facteur pronostic (génotype ?) Des traitements ciblant l’altération génétique ne peuvent pour le moment n’être proposé qu’aux patients présentant une altération génétique particulière.
 Des traitements ciblant l’altération génétique ne peuvent pour le moment n’être proposé qu’aux patients présentant une altération génétique particulière.")
63
Révision du diagnostic Un exemple
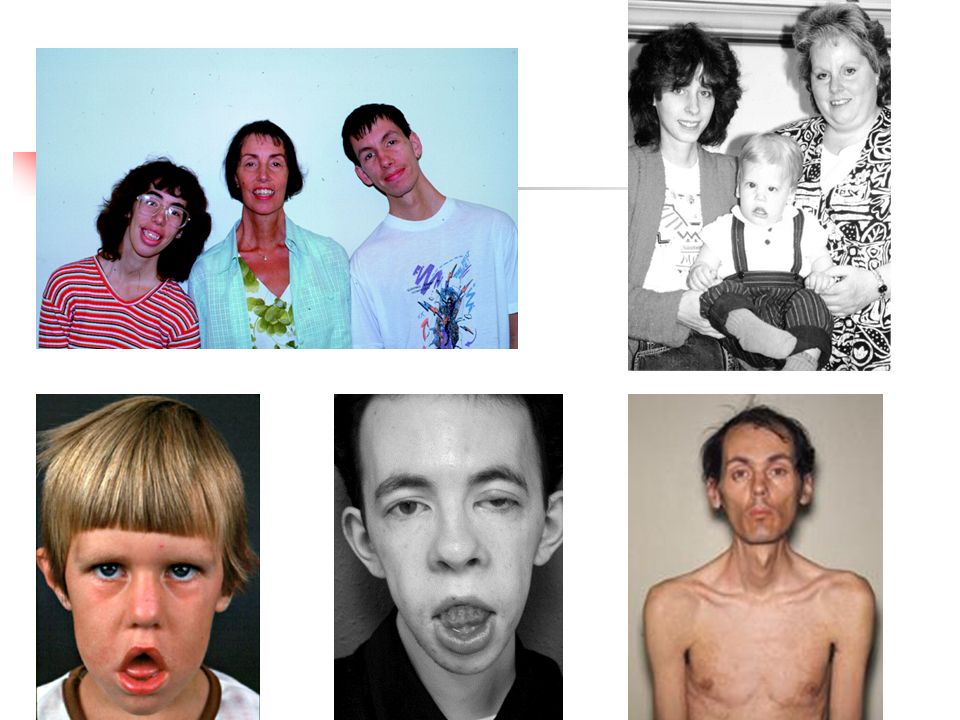
Homme sans antécédents familiaux En 1990 : A l’âge de 10 ans, diagnostic de dystrophie musculaire de Duchenne De 1990 à 2010 : Non suivi par une consultation spécialisée MNM En 2010 : A l’âge de 30 ans, marche encore possible avec appui, démarche dandinante, mais aucune atteinte cardiaque Le médecin myologue consulté, au vu de l'évolution, évoque une hypothèse diagnostique alternative ● Nouvelle biopsie musculaire en 2010 : - la dystrophine est de taille et de quantité normales - il ne peut donc s'agir d'une myopathie de Duchenne-Becker - on note en revanche l'absence d'une autre protéine membranaire, l’alpha-sarcoglycane ● Test génétique : mutation dans le gène de l’alpha-sarcocoglycane (LGMD2D) R77C On a révisé le diagnostic : Myopathie de Duchenne Alpha-sarcoglycanopathie
 R77C. On a révisé le diagnostic : Myopathie de Duchenne Alpha-sarcoglycanopathie.")
64
Conseil génétique si le diagnostic était celui de myopathie de Duchenne
Transmission récessive lié à l’X : Les garçons expriment la maladie Les femmes sont conductrices Le risque pour les apparentés est fonction de l’identification de la mutation chez la maman : - Si la maman est conductrice : Chaque sœur du patient a un risque de 50% d’être conductrice. Et si elle l’est, elle a un risque de 50% d’avoir un garçon malade. => Possibilité de réaliser un DPN - Si on ne retrouve pas la mutation chez la maman, il ne faut pas négliger le risque de mosaïque germinale

65
Nouveau Conseil génétique adapté au diagnostic d’Alpha-sarcoglycanopathie
Transmission autosomique récessive Touche autant garçons et filles. Malade =porteur de 2 copies non fonctionnelles du gène Parents = porteur d’une copie du gène non fonctionnel = hétérozygote Le risque pour les apparentés n’est pas un risque individuel mais c’est le risque pour un couple d’avoir un enfant atteint : 1- Déterminer le statut de l’apparenté : la mutation familiale 2- et celui du conjoint : la mutation familiale et la mutation la plus fréquente 3- Si ils sont tous les 2 porteurs: le risque est de 25% d’avoir un enfant atteint Mais le risque pour que le conjoint soit porteur d’une mutation est très faible et fonction de la fréquence de la maladie

Présentations similaires






>")
>")



>")
 Pr E. Tournier-Lasserve>")



 Pr E. Tournier-Lasserve>")

>")
>")



