Télécharger la présentation
1
La phylogénomique sans alignement de séquences
Jean-Loup Risler Statistique & Génome Carry-le-Rouet, Décembre 2006

2
Reconstructions phylogénétiques
Distances Maximum de parcimonie Maximum de vraisemblance Recherche de synténies Identification des orthologues Blast Alignements multiples

3
Une alternative: la recherche de « mots » communs
dans les séquences (k-words, k-tups, k-grams...) Exemple classique : le « Dotplot » « Dotplot » entre deux séquences génomiques de deux mycobactéries. Chaque croix indique un « mot » conservé de 20 lettres (au moins).
 Exemple classique : le « Dotplot » « Dotplot » entre deux séquences génomiques de deux mycobactéries. Chaque croix indique un « mot » conservé de 20 lettres (au moins).")
4
Les alignements de chromosomes entiers sont
impossibles à cause des réarrangements. La recherche de « mots communs » ne se soucie pas de leurs positions. On peut donc penser à déterminer une « distance » entre chromosomes à partir de leur contenu en mots communs.

5
Composition en di- et tri-nucléotides (chaînes de
B. E. Blaisdell, PNAS 83 (1986), Composition en di- et tri-nucléotides (chaînes de Markov d’ordre 1 et 2) de séquences codantes et non codantes test du chi2: S1 = mesure de la différence entre deux séquences basée sur leur composition en dinucléotides (chaînes de Markov d’ordre 1) S2 = mesure de la différence entre deux séquences basée sur leur composition en trinucléotides (chaînes de Markov d’ordre 2)
, Composition en di- et tri-nucléotides (chaînes de. Markov d’ordre 1 et 2) de séquences codantes et. non codantes test du chi2: S1 = mesure de la différence entre deux séquences basée sur leur composition en dinucléotides (chaînes de Markov d’ordre 1) S2 = mesure de la différence entre deux séquences basée sur leur composition en trinucléotides (chaînes de Markov d’ordre 2)")
6
5 B 4 3 2 Arbre obtenu par Baisdell à partir de la composition en dinucléotides des séquences de 30 gènes (exons) en 1986 A: vertébrés et B: invertébrés (sauf un vilain petit canard) A 1
 A. 1.")
7
{ Revue: Vinga, S. & Almeida, J. Alignment-free sequence comparison
Bioinformatics 19 (2003), Gary W. Stuart Une séquence peut être représentée par un vecteur AA TT AT { s 3 4 S = AATATTAAATTTATA AA = 3 AT = 4 TT = 3 TA = 4 s = (3, 4, 3, 4)
, Gary W. Stuart. Une séquence peut être représentée par un vecteur. AA. TT. AT. { s. S = AATATTAAATTTATA. AA = 3. AT = 4. TT = 3. TA = 4. s = (3, 4, 3, 4)")
8
{ 2 s1 TT { 4 s2 AA Une séquence S1 dupliquée en S2=S1-S1 seront représentées par deux vecteurs parallèles (angle nul) AT

9
{ { s1 s2 s1 . s2 = u1*u2 + v1*v2 + t1*t2 = |s1|*|s2|*cos()
AA AT s1 . s2 = u1*u2 + v1*v2 + t1*t2 = |s1|*|s2|*cos() |s1| = (u12 + v12 + t12)1/2 Deux séquences de compositions proches sont représentées par des vecteurs proches (petit angle) Chaque protéine est représentée par un vecteur dont les composantes (les compositions en mots) sont connues. L’angle entre les deux vecteurs se calcule très facilement.
 |s1| = (u12 + v12 + t12)1/2. Deux séquences de compositions proches sont représentées par des vecteurs proches (petit angle) Chaque protéine est représentée par un vecteur dont les composantes (les compositions en mots) sont connues. L’angle entre les deux vecteurs se calcule très facilement.")
10
d(i,j) = -Log[(1 + cos )/2]
Ici deux protéines sont représentées dans « l’espace des peptides ». Chaque protéine est caractérisée par sa composition en « peptides ». S’il s’agit de tétrapeptides, alors il y a 160,000 peptides différents (20^4), donc chaque protéine est représentée par un vecteur dans un espace à 160,000 dimensions. Deux protéines dont les compositions en tétrapeptides sont voisines seront représentées par deux vecteurs proches faisant entre eux un petit angle theta. On définit la « distance » d(i,j) entre deux protéines i et j à partir de l’angle que font leurs vecteurs. d(i,j) = -Log[(1 + cos )/2]
![d(i,j) = -Log[(1 + cos )/2]](http://slideplayer.fr/slide/1314496/3/images/10/d%28i%2Cj%29+%3D+-Log%5B%281+%2B+cos+%EF%81%B1%29%2F2%5D.jpg "Ici deux protéines sont représentées dans « l’espace des peptides ». Chaque protéine est caractérisée par sa composition en « peptides ». S’il s’agit de tétrapeptides, alors il y a 160,000 peptides. différents (20^4), donc chaque protéine est représentée par un vecteur dans un espace à 160,000 dimensions. Deux protéines dont les compositions en tétrapeptides sont voisines seront représentées. par deux vecteurs proches faisant entre eux un petit angle theta. On définit la « distance » d(i,j) entre deux protéines i et j à partir de l’angle que font leurs vecteurs. d(i,j) = -Log[(1 + cos )/2]")
11
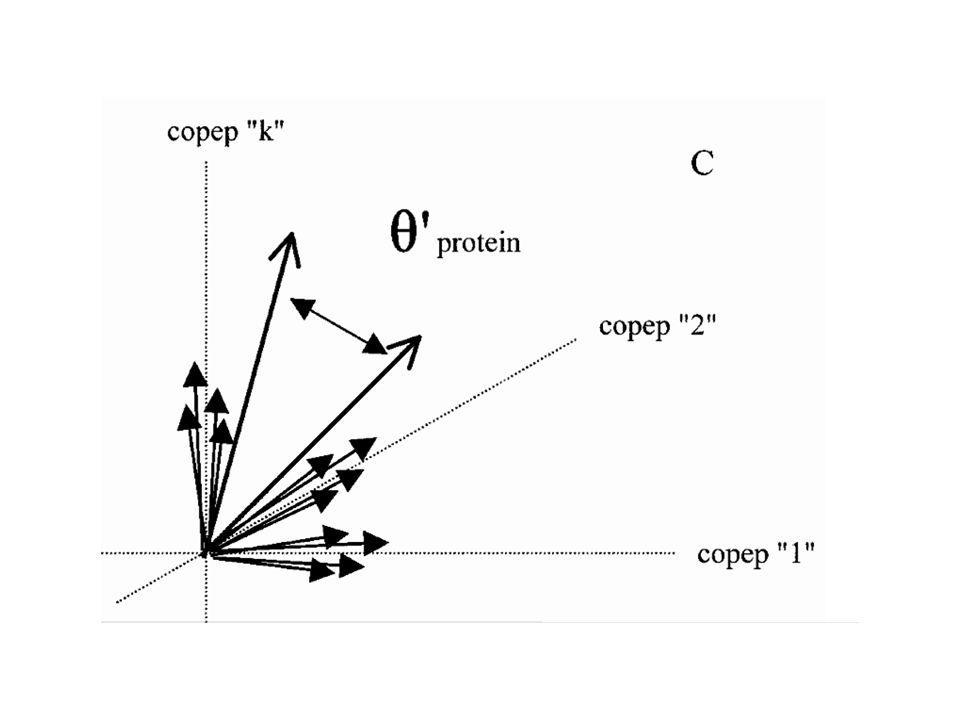
A l’inverse de la diapo précédente, nous sommes ici dans « l’espace des protéines ». Un tétrapeptide donné (par exemple KFGT) sera observé 2 fois dans la protéine 1, 3 fois dans la protéine 2, etc... Les tétrapeptides dont les fréquences sont voisines dans le jeu de protéines étudiées seront représentés par des vecteurs voisins.

12
Il y a 160.000 tetrapeptides possibles.
Donc, si l’on décompose un jeu de protéines en mots de longueur 4, chaque protéine sera représentée par un vecteur dans un espace à dimensions... De très nombreux « axes » portent peu d’information (tetrapeptides peu ou pas présents). On réduit la taille de l’espace en prenant pour repères les axes d’inertie les plus significatifs du nuage de points (changement de repère) et en supprimant les axes de faible inertie.
. On réduit la taille. de l’espace en prenant pour repères les axes d’inertie. les plus significatifs du nuage de points (changement. de repère) et en supprimant les axes de faible inertie.")
13
z B’ B’ A’ A B Un nuage de points a une forme d’ellipsoïde dans un espace à 3 dimensions x, y, z. On cherche à visualiser ce nuage de façon à ce qu’il soit le plus « éclaté » possible --> on détermine les axes d’inertie du nuage, on prend comme nouveau repère les 2 axes (A, A’) et (B,B’) qui portent la plus grande inertie et on projette le nuage sur leur plan. x A A’ y B
 et (B,B’) qui portent la plus grande inertie et on projette le nuage sur leur plan. x. A. A’ y. B.")
15
Toutes les protéines d’une même espèce sont regroupées
en un seul vecteur --> chaque espèce est représentée par un vecteur.

16
Arbre obtenu pour 64 espèces
Arbre obtenu pour 64 espèces. Cet arbre est le « consensus » des 17 arbres obtenus en réduisant l’espace des tetrapeptides ( dimensions) à dimensions.
 à dimensions.")


>")



 La phylogénie, ses concepts et ses applications à la génomiqueLa phylogénie, ses concepts et ses applications à la.>")
>")












>")