Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
C3b C3b Le complément S3 L2 Cédric Ménard 1

2
Définition du complément
Découverte scientifique (1895 Jules Bordet – Institut Pasteur- Nobel 1919) : - Sérum immun : bactéricide - Sérum immun chauffé : bactéries survivent - Sérum non-immun : bactéries survivent - Sérum immun chauffé + Sérum non-immun : bactéricide Injection bactéries Sérum Bactéries tuées Le sérum du lapin contient des Ac contre les bactéries qui les tuent Injection bactéries Ces Ac contre les bactéries ont besoin de molécules du sérum sensibles à la chaleur pour tuer les bactéries Bactéries vivent Bactéries tuées Les Ac ont besoin d’ un élément sensible à la chaleur et indpdt de l’Ag pour pouvoir tuer les bactéries… Qui est cet élément ? Les Ac dirigés contre les bactéries sont indispensables pour pouvoir les tuer Bactéries vivent
 : - Sérum immun : bactéricide. - Sérum immun chauffé : bactéries survivent. - Sérum non-immun : bactéries survivent. - Sérum immun chauffé + Sérum non-immun : bactéricide. Injection bactéries. Sérum. Bactéries tuées. Le sérum du lapin contient des Ac contre les bactéries qui les tuent. Injection bactéries. Ces Ac contre les bactéries ont besoin de molécules du sérum sensibles à la chaleur pour tuer les bactéries. Bactéries vivent. Bactéries tuées. Les Ac ont besoin d’ un élément sensible à la chaleur et indpdt de l’Ag pour pouvoir tuer les bactéries… Qui est cet élément Les Ac dirigés contre les bactéries sont indispensables pour pouvoir les tuer. Bactéries vivent.")
3
Définition du complément
Complément = élément thermolabile du sérum, non spécifique de l’antigène, capable de « complémenter » les Ac pour éliminer bactéries Branche de l’immunité innée Complément = ensemble de plus de 30 protéines : - plasmatiques (5% des protéines sériques) - de surface cellulaire Complément = ensemble de protéines
 - de surface cellulaire. Complément = ensemble de protéines.")
4
Définition du complément
Protéines du complément produites par : Foie (très majoritairement à l’état basal) Macrophages (production utile dans les OLS) Épithéliums muqueux et fibroblastes (faible) Inactif dans le sérum, clivages en cascade des zymogènes confèrent activités enzymatiques Deux types d’activateurs du complément : complexes immuns = complexes Ag / Ac (Ac-dépendant) sucres associés aux pathogènes (Ac-indépendant) Trois voies : classique (via Ac) des lectines (sans Ac) alterne (sans Ac) Finalité : assurer la mort du pathogène : DIRECTEMENT : rupture membranaire et choc osmotique INDIRECTEMENT : en l’opsonisant = facilitant sa reconnaissance par le SI (Gram + à paroi) Signaux de danger reconnus par le complément différents de ceux reconnus par la DC
 Macrophages (production utile dans les OLS) Épithéliums muqueux et fibroblastes (faible) Inactif dans le sérum, clivages en cascade des zymogènes confèrent activités enzymatiques. Deux types d’activateurs du complément : complexes immuns = complexes Ag / Ac (Ac-dépendant) sucres associés aux pathogènes (Ac-indépendant) Trois voies : classique (via Ac) des lectines (sans Ac) alterne (sans Ac) Finalité : assurer la mort du pathogène : DIRECTEMENT : rupture membranaire et choc osmotique. INDIRECTEMENT : en l’opsonisant = facilitant sa reconnaissance par le SI (Gram + à paroi) Signaux de danger reconnus par le complément différents de ceux reconnus par la DC.")
5
I- Nomenclature Fragments de clivage enzymatique : lettre minuscule «C4 clivé donne C4a + C4b » Molécule inactive : désignée par i « C3bi ou iC3b » Formes enzymatiques actives : recouvertes d’une barre horizontale « C1r »

6
II- Les voies d’activation
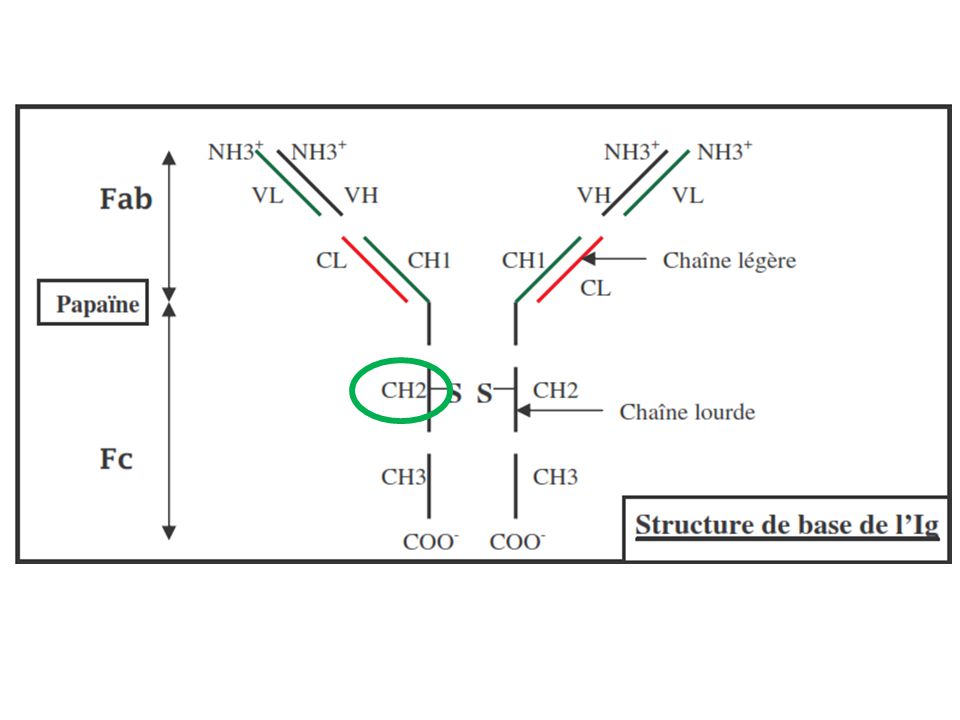
La présence d’Ac spécifiques indique qu’une réponse contre l’Ag a déjà été initiée ! A- La voie classique (nécessite Ac) C1q C1r C1s C1-estérase (Ca2+-dpdte) sérine-protéases C1r et C1s activées IgG (IgG1 et IgG3) IgM IgG IgM Surface de la cible bactérie par exemple C1 se fixe par C1q (protéine de reconnaissance) sur le Fc des anticorps impliqué dans des complexes immuns (région CH2 pour IgG1 et IgG3 et région CH4 pour IgM) Activation par des complexes immuns à IgM pentamérique (3 sites pour C1) ou IgG (1 site pour C1 et donc plusieurs IgG nécessaires) IgM 1000 x plus efficaces que les IgG pour activer C1 (passage forme plane à agrafe) CH=CDR
 C1q. C1r. C1s. C1-estérase (Ca2+-dpdte) sérine-protéases C1r et C1s activées. IgG. (IgG1 et IgG3) IgM. IgG. IgM. Surface de la cible. bactérie par exemple. C1 se fixe par C1q (protéine de reconnaissance) sur le Fc des anticorps impliqué dans des complexes immuns (région CH2 pour IgG1 et IgG3 et région CH4 pour IgM) Activation par des complexes immuns à IgM pentamérique (3 sites pour C1) ou IgG (1 site pour C1 et donc plusieurs IgG nécessaires) IgM 1000 x plus efficaces que les IgG pour activer C1 (passage forme plane à agrafe) CH=CDR.")
8
II- Les voies d’activation
A- La voie classique 1- Activation C2b covalent C4a Fixation C1 à l’anticorps via C1q Activation autocatalytique du proenzyme C1r en C1r C1r convertit C1s en C1s C1s va cibler C4 circulant C4a + C4b (C4b s’ancre dans la membrane cible via thioester R-S-CO-R’) C1s va cibler C2 associé à C4b C2a +C2b C2a s’associe à C4b déjà ancré dans la membrane et donne C4b2a (=C4bC2a) Ce complexe C4b2a = C3 convertase « classique » qui clive C3 en C3a + C3b (expose un thioester)
 C1s va cibler C2 associé à C4b C2a +C2b. C2a s’associe à C4b déjà ancré dans la membrane et donne C4b2a (=C4bC2a) Ce complexe C4b2a = C3 convertase « classique » qui clive C3 en C3a + C3b (expose un thioester)")
9
C1-inh (détache C1r et C1s du complexe)
II- Les voies d’activation A- La voie classique 2- Régulation Contrôle de la C1-estérase par C1-inh : - empêche activation de C1r en phase liquide - peut inactiver le C1r activé Thioester C3b et C4b hyper-réactif est hydrolysé en qqes ms par l’eau si non-fixé = obligation de se lier covalement au voisinage immédiat (= pas d’effet collatéral) Complexe C4b2a instable = dissociation spontanée Contrôle de la C3 convertase (C4b2a) : - C4-bp inhibe l’association C4b / C2 - CD55 = DAF « decay-accelerating factor » dissociation C4 / C2 (protection de l’hôte) - CR1 (CD35) = C3b-R peut titrer le C3b - Facteur I : clive C3b en iC3b
 Complexe C4b2a instable = dissociation spontanée. Contrôle de la C3 convertase (C4b2a) : - C4-bp inhibe l’association C4b / C2. - CD55 = DAF « decay-accelerating factor » dissociation C4 / C2 (protection de l’hôte) - CR1 (CD35) = C3b-R peut titrer le C3b. - Facteur I : clive C3b en iC3b.")
10
Reconnaît les sucres des pathogènes différents de ceux de l’hôte
II- Les voies d’activation B- La voie des lectines : Ficoline et MBL (court-circuitent les Ac) Reconnaît les sucres des pathogènes différents de ceux de l’hôte Ici la protéine de reconnaissance est MBL Activation : Mannan Binding Lectine (MBL) ou Ficoline reconnaissant sucres des pathogènes (mécanisme analogue à la C1 estérase qui reconnaît les complexes immuns) Molécules effectrices : MBL-associated proteases = MASP (homologues de C1r/s)
 Reconnaît les sucres des pathogènes différents de ceux de l’hôte. Ici la protéine de reconnaissance est MBL. Activation : Mannan Binding Lectine (MBL) ou Ficoline reconnaissant sucres des pathogènes (mécanisme analogue à la C1 estérase qui reconnaît les complexes immuns) Molécules effectrices : MBL-associated proteases = MASP (homologues de C1r/s)")
11
II- Les voies d’activation
Ac-indépendante ! II- Les voies d’activation B- La voie des lectines (Ficoline et MBL) 1- Activation MBL ou ficoline fixe sucres cibles = MASP clivées / réarrangées et autoactivées (Ca 2+-dpdt) MASP1 et 2 clivent C4 en C4a + C4b (qui s’ancre dans membrane) MASP1 et 2 clivent C2 associé à C4b membranaire en C2a pour donner C4b2a Formation de C4b2a = C3 convertase qui donne C3a + C3b (qui s’ancre dans membrane) MASP3 (splicing alternatif de MASP1) : rôle encore flou
 1- Activation. MBL ou ficoline fixe sucres cibles = MASP clivées / réarrangées et autoactivées (Ca 2+-dpdt) MASP1 et 2 clivent C4 en C4a + C4b (qui s’ancre dans membrane) MASP1 et 2 clivent C2 associé à C4b membranaire en C2a pour donner C4b2a. Formation de C4b2a = C3 convertase qui donne C3a + C3b (qui s’ancre dans membrane) MASP3 (splicing alternatif de MASP1) : rôle encore flou.")
12
II- Les voies d’activation
C1-inh II- Les voies d’activation B- La voie des lectines (Ficoline et MBL) 2- Régulation C3b doit se fixer à un groupe hydroxyl (-OH) de son voisinage immédiat (sucre / prot memb) Si absence de fixation, molécule H2O fixe C3b = ne peut plus lier une membrane cellulaire (C3b très instable) « But : éviter accumulation de C3b actif qui pourrait être délétère pour l’hôte (vrai aussi pour le C3b issu de la voie classique) » MBL : forte affinité pour sucres bactériens, levures, virus : Mannose N-acétyl-glycosamine Fucose Mais MBL peu affine pour sucres de mammifères (propriétés stériques des sucres) : Acide sialique Galactose Concentration de l’activité du C3b à la surface membranaire
 2- Régulation. C3b doit se fixer à un groupe hydroxyl (-OH) de son voisinage immédiat (sucre / prot memb) Si absence de fixation, molécule H2O fixe C3b = ne peut plus lier une membrane cellulaire (C3b très instable) « But : éviter accumulation de C3b actif qui pourrait être délétère pour l’hôte (vrai aussi pour le C3b issu de la voie classique) » MBL : forte affinité pour sucres bactériens, levures, virus : Mannose. N-acétyl-glycosamine. Fucose. Mais MBL peu affine pour sucres de mammifères (propriétés stériques des sucres) : Acide sialique. Galactose. Concentration de l’activité du C3b à la surface membranaire.")
13
II- Les voies d’activation
« L'hydrolyse d'une substance est sa décomposition par l'eau grâce aux ions H3O+ et HO- provenant de la dissociation de l'eau » II- Les voies d’activation C- La voie alterne (80% de l’activation du Cplmt) « La voie tourne au ralenti » Hydrolyse lente dans plasma C3 C3b C3b C2a = enzyme hydrolytique (estérase) donc catalyse l’hydrolyse de C3b (plus rapide) Initiation indépendante d’activateurs = clivage spontané / lent de C3 à bas bruit en C3a + C3b Phase d’amplification par des sucres bactériens (LPS, bactéries Gram+) ou des virus = déclenchement réel, « explosion » de la voie La cascade s’amplifie sur les cellules étrangères mais est immédiatement régulée sur les cellules saines de l’hôte
 « La voie tourne au ralenti » Hydrolyse lente dans plasma. C3. C3b. C3b. C2a = enzyme hydrolytique (estérase) donc catalyse l’hydrolyse de C3b (plus rapide) Initiation indépendante d’activateurs = clivage spontané / lent de C3 à bas bruit en C3a + C3b. Phase d’amplification par des sucres bactériens (LPS, bactéries Gram+) ou des virus = déclenchement réel, « explosion » de la voie. La cascade s’amplifie sur les cellules étrangères mais est immédiatement régulée sur les cellules saines de l’hôte.")
14
II- Les voies d’activation
C- La voie alterne C3b protéase C3b 1- Activation C3 hydrolysé à bas bruit et donne C3b Liaison facteur B à C3b (Mg2+- dpdt) C3bB est reconnu et clivé par la protéase « facteur D » Clivage de C3bB en C3bBb = C3 convertase alterne C3bBb clive C3 en C3a et C3b = constituant de la C3 convertase : AMPLIFICATION La cible est « recouverte » par de nbreuses molécules de C3b (opsonisation) Les C3b peuvent former de nouvelles C3bB Stabilisation du C3bBb par la properdine (P) liant C3b (demi-vie 90 secondes vs 30 minutes), la properdine reconnaît les PAMP et les DAMP = des marqueurs de danger L’hydrolyse du C3 donne du C3b comme ds les autres voies
 C3bB est reconnu et clivé par la protéase « facteur D » Clivage de C3bB en C3bBb = C3 convertase alterne. C3bBb clive C3 en C3a et C3b = constituant de la C3 convertase : AMPLIFICATION. La cible est « recouverte » par de nbreuses molécules de C3b (opsonisation) Les C3b peuvent former de nouvelles C3bB. Stabilisation du C3bBb par la properdine (P) liant C3b (demi-vie 90 secondes vs 30 minutes), la properdine reconnaît les PAMP et les DAMP = des marqueurs de danger. L’hydrolyse du C3 donne du C3b comme ds les autres voies.")
15
II- Les voies d’activation
C- La voie alterne 2- Régulation Facteur H : dissociation C3bBb Facteur I : en association avec le CD46 (=MCP : membrane cofactor Protein), clivage C3b en C3bi, = le facteur B ne peut plus se lier à C3b devenu C3bi « De façon générale, dissociation des complexes de convertases est irréversible et permet de down-réguler la réponse du complément » C3b est au coeur de plusieurs voie d’activation/régulation
, clivage C3b en C3bi, = le facteur B ne peut plus se lier à C3b devenu C3bi. « De façon générale, dissociation des complexes de convertases est irréversible et permet de down-réguler la réponse du complément » C3b est au coeur de plusieurs voie d’activation/régulation.")
16
Récapitulatif C3b C3b En dépit d’initiateurs différents, les 3 voies convergent vers une C3 convertase : C4b2a pour la voie classique et la voie des lectines C3bBb pour la voie alterne

17
III- De la C3 convertase au complexe d’attaque membranaire :
III- De la C3 convertase au complexe d’attaque membranaire : la voie effectrice commune Voie classique Voie des lectines Voie alterne C3 convertases C5 convertases Un fragment C3b s’associe aux C3 convertases et forme : C4b2a3b C3bBb3b Les C5 convertases permettant de cliver C5 en C5a + C5b (solubles)
")
18
III- De la C3 convertase au complexe d’attaque membranaire :
III- De la C3 convertase au complexe d’attaque membranaire : la voie effectrice commune C5b soluble s’associe à C6 et C7, le complexe hydrophobe ainsi formé s’insère dans la membrane cible via C7 C5b67 ancré dans une membrane = récepteur à C8 ce qui aboutit à C5b678 qui initie une réaction lente de lyse C9 (x environ 12) est recruté au complexe membranaire C5b678 pour constituer le MAC « membrane-attack complex »
 est recruté au complexe membranaire C5b678 pour constituer le MAC « membrane-attack complex »")
19
III- De la C3 convertase au complexe d’attaque membranaire :
III- De la C3 convertase au complexe d’attaque membranaire : la voie effectrice commune C9 Composition du MAC hétérogène : C5b6789n , n = 1 à 12 10 nm Perte de l’équilibre osmotique (l’eau rentre dans la cellule/le pathogène) Mort mécanique de la cible Gram-negative bacterium Shigella dysenteriae
 Mort mécanique de la cible. Gram-negative bacterium Shigella dysenteriae")
20
III- De la C3 convertase au complexe d’attaque membranaire :
III- De la C3 convertase au complexe d’attaque membranaire : la voie effectrice commune Régulation Régulation des voies d’activation est majoritaire mais la voie commune est aussi régulée : C5b67 doit s’ancrer très vite dans la cible, sinon site d’ancrage inactivé par hydrolyse par fixation à protéines plasmatiques (protéine S = vitronectine ou clusterine) C8 en se liant à C5b67 en phase liquide bloque le site d’ancrage dans la membrane Plupart des membranes de l’hôte expriment CD59 qui se loge dans le CAM en cours d’assemblage = empêche la liaison de C9 = empêche formation du pore
 C8 en se liant à C5b67 en phase liquide bloque le site d’ancrage dans la membrane. Plupart des membranes de l’hôte expriment CD59 qui se loge dans le CAM en cours d’assemblage = empêche la liaison de C9 = empêche formation du pore.")
21
IV- Le complément dans l’Évolution

22
V- Récepteurs cellulaires des composants du complément
« ancrage » A- Récepteur fragments « majeurs » CR1 = CD35 : Récepteur du C3b et du C4b (GR, Leucocytes, FDC) Cibles opsonisées ou complexes immuns (GR) puis phagocytose par macrophages du foie (cellules de Kupffer) Rôle d’atténuation en liant C3b et C4b CR2 = CD21 : Récepteur de C3d (Lymphos B, FDC) Reconnaissance des lymphos B par virus EBV CD21 diminue seuil d’activation BCR et favorise survie B « fixation à un R » Les fragments de clivage de C3 (C3b, iC3b, C3d) Reconnaissance via BCR Activation + Fragments iC3b = C3d B B Reconnaissance via BCR + CR2 Activation +++ (1000x plus efficace)
 Cibles opsonisées ou complexes immuns (GR) puis phagocytose par macrophages du foie (cellules de Kupffer) Rôle d’atténuation en liant C3b et C4b. CR2 = CD21 : Récepteur de C3d (Lymphos B, FDC) Reconnaissance des lymphos B par virus EBV. CD21 diminue seuil d’activation BCR et favorise survie B. « fixation à un R » Les fragments de clivage de C3 (C3b, iC3b, C3d) Reconnaissance via BCR. Activation + Fragments iC3b = C3d. B. B. Reconnaissance via BCR + CR2. Activation +++ (1000x plus efficace)")
23
V- Récepteurs cellulaires des composants du complément
B- Récepteur fragments « mineurs » CR3 = CD11b/CD18 : Récepteur iC3b (Majorité des leucocytes) Molécule d’adhérence Opsonisation / phagocytose bactéries ou cellules recouvertes de iC3b Facilitation des contacts cellulaires CR4 = CD11c/CD18 : Récepteur iC3b (Cellules myéloïdes et certains lymphocytes activés) Rôle physiologique peu connu C- Autres récepteurs (récepteurs à protéine G) C3aR (éosninophiles, basophiles et neutrophiles) C5aR (leucocytes et qqes autres types cellulaires) C1qR
 Molécule d’adhérence. Opsonisation / phagocytose bactéries ou cellules recouvertes de iC3b. Facilitation des contacts cellulaires. CR4 = CD11c/CD18 : Récepteur iC3b (Cellules myéloïdes et certains lymphocytes activés) Rôle physiologique peu connu. C- Autres récepteurs (récepteurs à protéine G) C3aR (éosninophiles, basophiles et neutrophiles) C5aR (leucocytes et qqes autres types cellulaires) C1qR.")
24
VI- Les autres rôles du complément
Outre son rôle de lyse, le complément intervient dans : - l’opsonisation des pathogènes / C apoptotiques (perte CD46 et CD59) - l’activation de la réponse inflammatoire Ceci est permis par les 4 récepteurs aux fragments C3 du complément A- L’opsonisation cible Reconnaissance accrue via CRs Phagocytose 100 x plus efficace phagocyte cible phagocyte Reconnaissance moyenne cible phagocyte Reconnaissance accrue via les R-Fc Rôle capital du complément ds la “vie de ts les jours” (cell apoptotiques) et la phagocytose rapide (avant action des lymphocytes) cf neutrophiles et la NFS cible phagocyte Reconnaissance « maximale » via R-Fc + CR En facilitant la reconnaissance de l’Ag par les futures APC, le complément fait le lien immunité innée / adaptative cellulaire
 - l’activation de la réponse inflammatoire. Ceci est permis par les 4 récepteurs aux fragments C3 du complément. A- L’opsonisation. cible. Reconnaissance accrue via CRs Phagocytose 100 x plus efficace. phagocyte. cible. phagocyte. Reconnaissance moyenne. cible. phagocyte. Reconnaissance accrue via les R-Fc. Rôle capital du complément ds la vie de ts les jours (cell apoptotiques) et la phagocytose rapide (avant action des lymphocytes) cf neutrophiles et la NFS. cible. phagocyte. Reconnaissance « maximale » via R-Fc + CR. En facilitant la reconnaissance de l’Ag par les futures APC, le complément fait le lien immunité innée / adaptative cellulaire.")
25
VI- Les autres rôles du complément
B- L’activation de la réponse inflammatoire Les fragments de clivage C3a, C4a et C5a du compléments ne sont pas inactifs ! Ces molécules sont nommées « anaphylatoxines » et permettent : - d’augmenter la perméabilité vasculaire (rôle dans l’extravasation) - d’activer la dégranulation des polynucléaires, mastocytes (histamine…), éosinoph - la contraction des muscles lisses - le chimiotactisme des cellules de l’inflammation : C5a attire les MΦ sur site inflammation et facilite ainsi la phagocytose des cibles (opsonines et cplmt-R) - d’augmenter l’expression des FcgR des MΦ (C5a)
 - d’activer la dégranulation des polynucléaires, mastocytes (histamine…), éosinoph. - la contraction des muscles lisses. - le chimiotactisme des cellules de l’inflammation : C5a attire les MΦ sur site inflammation et facilite ainsi la phagocytose des cibles (opsonines et cplmt-R) - d’augmenter l’expression des FcgR des MΦ (C5a)")
26
RCA = régulateurs de l’activation du complément (ex : DAF=CD55)
VII- Échappement au complément RCA = régulateurs de l’activation du complément (ex : DAF=CD55) Produits par l’hôte, ils peuvent être capturés à la surface du pathogène par des protéines spécialisées Protéine virale DAF humain Gène viraux peuvent coder pour des analogues de RCA ! « mimétisme moléculaire » Recrutement / mimicking des régulateurs du complément - Modulation du complément (CD59-like, Ig inactivating-proteine…) Dégradation enzymatique : bactéries uniquement (C1q, Ac, C5a, P…) Evasion passive : paroi bactérienne des Gram + !
 Produits par l’hôte, ils peuvent être capturés à la surface du pathogène par des protéines spécialisées. Protéine virale. DAF humain. Gène viraux peuvent coder pour des analogues de RCA ! « mimétisme moléculaire » Recrutement / mimicking des régulateurs du complément. - Modulation du complément (CD59-like, Ig inactivating-proteine…) Dégradation enzymatique : bactéries uniquement (C1q, Ac, C5a, P…) Evasion passive : paroi bactérienne des Gram + !")
27
VIII- Pathologies associées au complément
Maladies génétiques liées à un déficit en complément sont rares : déficits protéines de la voie classique Infections récurrentes (méningites, pneumonies…) Association fréquente au lupus déficits protéines de la voie lectines et alterne : infection récurrentes déficits protéines du MAC accompagnés d'infections à Neisseria meningitidis déficits inhibiteurs donnent infections et des MAI Cf clearance des corps apoptotiques et lupus
 Association fréquente au lupus. déficits protéines de la voie lectines et alterne : infection récurrentes. déficits protéines du MAC accompagnés d infections à Neisseria meningitidis. déficits inhibiteurs donnent infections et des MAI. Cf clearance des corps apoptotiques et lupus.")
28
VIII- Pathologies associées au complément
HNP : Hémoglobinurie Paroxystique Nocturne Globules rouges plus sensibles à la CDC suite à une mutation génétique rendant CD59 non fonctionnel Hémolyse : anémie, toxicité rénale Eculizumab : IgG4 Bloque le site de clivage de C5 (C5a) Cf clearance des corps apoptotiques et lupus
 Cf clearance des corps apoptotiques et lupus.")
29
Lymphocytes Natural Killer (NK) et T
S3 L2 Cédric Ménard

30
Cinétique de la réponse immunitaire
Anticorps T8 cytotoxiques NK T IFN de type I

31
Caractéristiques de l ’immunité innée et de l ’immunité acquise
Mécanismes de reconnaissance de l ’immunité innée réponse rapide (heures) invariable nombre limité de spécificités constant lors de la réponse Mécanismes de reconnaissance de l ’immunité adaptative réponse lente (jours ou semaines) variable nbreuses spécificités hautement sélectives s ’améliore lors de la réponse Mécanismes effecteurs communs pour la destruction du pathogène
 invariable. nombre limité de spécificités. constant lors de la réponse. Mécanismes de reconnaissance. de l ’immunité adaptative. réponse lente (jours ou semaines) variable. nbreuses spécificités hautement sélectives. s ’améliore lors de la réponse. Mécanismes effecteurs communs pour la destruction du pathogène.")
32
Les cellules Natural Killer « NK »

33
I- Caractéristiques générales
Grand lymphocyte granuleux Fait partie de l’immunité innée : peut détruire rapidement des cellules tumorales ou infectées sans activation ni prolifération préalable Distribution dans l’organisme : Sang : 5-15% des lymphocytes circulant Organes lymphoïdes : ganglions, rate, amygdales Tissus périphériques : foie, poumon (« sentinelles »), endomètre Expriment CD56, CD16, et NKp46 (pas CD3 ni CD19) Production dans la moelle, maturation dans les organes lymphoïdes (ganglions ++) Lymphocytes cytotoxiques mais aussi producteurs de cytokines/chimiokines Lysent sans immunisation à la différence des T CD8
, endomètre. Expriment CD56, CD16, et NKp46 (pas CD3 ni CD19) Production dans la moelle, maturation dans les organes lymphoïdes (ganglions ++) Lymphocytes cytotoxiques mais aussi producteurs de cytokines/chimiokines. Lysent sans immunisation à la différence des T CD8.")
34
II- Ontogénèse et maturation des NK
NK matures Progéniteur Lymphoïde Commun NK immature NK CD56 high NK CD56 faible Progéniteur NK CSH E4BP4 Id2 Production majoritaire par la moelle, maturation dans les ganglions Les NK CD56 faible vont retourner dans le sang sang B moelle Ganglions (paracortex)
")
35
II- Ontogénèse et maturation des NK
Rôle central de l’IL-15 dans toutes les étapes de la lymphopoïèse NK Produite par les cellules stromales dans la moelle Produite par les DC/macrophages dans les ganglions “Education” des NK au cours de leur maturation Doivent rencontrer un CMH-I au cours de leur développement Nécessaire pour acquérir leur fonctionnalité KIR Maturation finale Potentiel cytotoxique et de sécrétion de cytokines NK immature KIR : Killer cell Immunoglobulin-like Receptor Anergie CMH-I

36
2 sous-types de NK matures
CD56high - Dans les ganglions (CCR7+) et la muqueuse utérine - Peu cytotoxiques - Produisent beaucoup de cytokines CD56low - Dans le sang principalement Cytotoxiques (perforine ds granules préformés) Produisent moins de cytokines Récepteurs aux chimiokines : migration dans les tissus surtout si inflammation
 et la muqueuse utérine. - Peu cytotoxiques. - Produisent beaucoup de cytokines. CD56low. - Dans le sang principalement. Cytotoxiques (perforine ds granules préformés) Produisent moins de cytokines. Récepteurs aux chimiokines : migration dans les tissus surtout si inflammation.")
37
III- Reconnaissance de la cible
Cellules tueuses III- Reconnaissance de la cible T8 NK R act R inh CMH-I CMH-I Pas restreinte à un complexe CMH-peptide Résulte d’une balance entre : Signaux inhibiteurs (tolérance au soi) : KIR inhibiteur vs CMH-I Signaux activateurs : ligand = non soi/soi altéré Répertoire NK : Chaque individu diffère dans ses récepteurs KIR exprimé par ses NK (gènes polymorphiques) Chaque NK a une combi ≠ de R inh et la plupart des NK a au moins 1 R inh (sinon, ils sont anergiques) Existence d’une tolérance au soi = « nos propres NK ne nous attaquent pas » cible cible Pas de balance pour le TCD8 : présentation du peptide ds du CMH I et c’est tout
 : KIR inhibiteur vs CMH-I. Signaux activateurs : ligand = non soi/soi altéré. Répertoire NK : Chaque individu diffère dans ses récepteurs KIR exprimé par ses NK (gènes polymorphiques) Chaque NK a une combi ≠ de R inh et la plupart des NK a au moins 1 R inh (sinon, ils sont anergiques) Existence d’une tolérance au soi = « nos propres NK ne nous attaquent pas » cible. cible. Pas de balance pour le TCD8 : présentation du peptide ds du CMH I et c’est tout.")
38
Les récepteurs activateurs du NK
Exprimés constitutivement à la membrane des NK, reconnaissent des motifs moléculaires conservés NKG2D (lectine de type C): cytotoxicité naturelle Ligands : MICA et B, ULBP exprimés à la membrane lors d’un stress cellulaire : infection virale, transformation tumorale, lésions de l’ADN… Natural Cytotoxicity Receptor (NKp30,44,46) : cytox nat. Ligands : protéines virales (hémagglutinine ou pp65 du CMV) exprimées à la surface des cellules infectées CD16 (FcgRIIIa) : cytotoxicité dépendante des anticorps Ligand : cible reconnue par Ac qui se fixe sur son Ag spécifique CD16 se fixe à la partie Fc de l’Ac Autres : NKp44, DNAM-1, NKp80, 2B4…
: cytotoxicité naturelle. Ligands : MICA et B, ULBP exprimés à la membrane lors d’un stress cellulaire : infection virale, transformation tumorale, lésions de l’ADN… Natural Cytotoxicity Receptor (NKp30,44,46) : cytox nat. Ligands : protéines virales (hémagglutinine ou pp65 du CMV) exprimées à la surface des cellules infectées. CD16 (FcgRIIIa) : cytotoxicité dépendante des anticorps. Ligand : cible reconnue par Ac qui se fixe sur son Ag spécifique. CD16 se fixe à la partie Fc de l’Ac. Autres : NKp44, DNAM-1, NKp80, 2B4…")
39
Les récepteurs inhibiteurs du NK
KIR : Killer cell Immunoglobulin-like Receptor Famille de protéines codées par plusieurs gènes polymorphiques Diffèrent par le nbre de Domaines Ig extracellulaires (2D ou 3D) et la taille de leur queue cytoplasmique (Long ou Short) Les KIR2DL ou KIR3DL donnent un signal inhibiteur au NK Ligand CMH-I spécifique pour chaque KIR2DL ou KIR3DL : NKG2A : lectine de type C Association obligatoire avec le co-récepteur CD94 Ligand : CMH-I non classique : HLA-E KIR2DL1, 2 et 3 HLA-C KIR2DL4 HLA-G KIR3DL1 HLA-B KIR3DL2 HLA-A Tous les NK fonctionnels ont au moins 1 KIRDL donc ne vont pas s’attaquer au soi
 et la taille de leur queue cytoplasmique (Long ou Short) Les KIR2DL ou KIR3DL donnent un signal inhibiteur au NK. Ligand CMH-I spécifique pour chaque KIR2DL ou KIR3DL : NKG2A : lectine de type C. Association obligatoire avec le co-récepteur CD94. Ligand : CMH-I non classique : HLA-E. KIR2DL1, 2 et 3. HLA-C. KIR2DL4. HLA-G. KIR3DL1. HLA-B. KIR3DL2. HLA-A. Tous les NK fonctionnels ont au moins 1 KIRDL donc ne vont pas s’attaquer au soi.")
40
Les Récepteurs inhibiteurs Les Récepteurs activateurs
IgG fixé à une cellule Molécules du stress cellulaire : - MICA - MICB - ULBP CMH-I Protéines virales : pp65 (NKp30) hémagglutinine (NKp46) CMH-I type E KIR NCR NKG2D CD16 NKG2A CD94 ITIM ITAM et ITIM Protéines virales : hémagglutinine de influenza virus par ex (grippe et NK ds le poumon) ITAM Signal activateur : ITAM recrutent des kinases (immune receptor tyrosine-based activation motif) Signal inhibiteur : ITIM recrute phosphatases (immune receptor tyrosine-based inhibitory motif)
 hémagglutinine (NKp46) CMH-I type E. KIR. NCR. NKG2D. CD16. NKG2A. CD94. ITIM. ITAM et ITIM. Protéines virales : hémagglutinine de influenza virus par ex (grippe et NK ds le poumon) ITAM. Signal activateur : ITAM recrutent des kinases (immune receptor tyrosine-based activation motif) Signal inhibiteur : ITIM recrute phosphatases (immune receptor tyrosine-based inhibitory motif)")
41
Balance entre signaux activateurs et inhibiteurs
IgG Cytokines activatrices : IL IFN CD16 Ag + NCR prot. virales + + + MICA/B ULBP NKG2D NKG2A HLA-E - Attention le KIR n’est pas spécifique d’un antigène mais d’une molécule de CMH Attention les prot virales sont entières. Ce ne sont pas des peptides présentés ds du CMH I. rappel : NK n’a pas de TCR. Ses récepteurs activateurs ne reconnaissent qu’un tout petit répertoire de ligand conservé - Cytokines inhibitrices : TGF - cellule cible KIR NK HLA-A,B,C,G

42
Balance des signaux act et inh
« Ce n'est pas l'absence de molécules du CMH de classe I qui déclenche l'activation des lymphocytes NK mais la présence de ligands activateurs non compensée par des signaux inhibiteurs suffisants » Balance des signaux act et inh Ainsi, l'absence de molécules du CMH-I ne suffit pas à rendre certaines cellules sensibles à la lyse NK, par exemple : globules rouges neurones hépatocytes NK Les souris/patients mutés pour leur CMH1 ne font pas de MAI dues aux NK

43
i a a Une cellule du soi est normalement protégée de la lyse NK car le signal inhibiteur l’emporte en général sur l’activateur ! Une cellule « suspecte » (qui n’exprime pas de CMH-I) est lysée car aucun signal inhibiteur ne vient contrebalancer l’activateur d Hyperactivation NK Lyse possible a cytokines i + a a a a a a CD16 Une cellule allogénique est lysée car le CMH-I du non-soi ne déclenche pas de signal inhibiteur Une forte activation des NK peut contrebalancer les signaux inhibiteurs (ADCC, IL-2…)
 est lysée car aucun signal inhibiteur ne vient contrebalancer l’activateur. d Hyperactivation NK. Lyse possible. a. cytokines. i. + a. a. a. a. a. a. CD16. Une cellule allogénique est lysée car le CMH-I du non-soi ne déclenche pas de signal inhibiteur. Une forte activation des NK peut contrebalancer les signaux inhibiteurs (ADCC, IL-2…)")
44
IV- Mécanismes effecteurs
NK IV- Mécanismes effecteurs Les signaux activateurs l’emportent… que se passe-t-il ? 1 - Lyse de la cible selon 2 systèmes : - Perforine-granzyme - Récepteurs de mort : Fas/FasL, TRAIL/TRAIL-R 2 - Synthèse de cytokines : TNFa, IFNg, GM-CSF - Orientation Th1 - Contrôle direct de la réplication virale par l’ IFNg - Activation MΦ et des DC 3 - Synthèse de chimiokines inflammatoires : CXCL8, CCL3, CCL4, CCL5 donc recrutement de : - PNN - Macrophages - DC immatures - T activés ? Lien entre l’inné et l’adaptatif

45
Voie perforine/granzyme Voie des récepteurs de mort
Cytotoxicité NK Voie perforine/granzyme Voie des récepteurs de mort TRAIL FasL TNF Perforine Granzyme B Fas TNFR-I TRAIL-R Apoptose de la cible

46
Voie extrinsèque + BID clive Voie intrinsèque apoptosome NOYAU

47
V-Explications moléculaires de la réponse NK
-1- Reconnaissance allogénique : - fondée sur la différence d’expression « KIR de l’hôte / CMH-I du donneur » = les CMH-I de l’hôte n’induisent pas le signal inhibiteur chez la NK qui ne les reconnaît pas. - Implication dans la greffe en général NK cible R act R inh Toutes les combinaisons ne sont pas possibles !!! Receveur Donneur HLA très polymorphique donc pas les mêmes HLA A B C d’un individu à l’autre

48
V-Explications moléculaires de la réponse NK
- 2 - Réponse anti-tumorale : Pertes d’expression classe I sur 80% tumeurs (pas de signal inhibiteur, échappement tumoral) Ligands des Récepteurs activateurs surexprimés en condition de stress (en surnombre ils déséquilibrent la balance vers l’activation) NK Cancer epithélial = carcinome ULBP : UL16-binding protein MIC : MHC I polypeptide related
 Ligands des Récepteurs activateurs surexprimés en condition de stress (en surnombre ils déséquilibrent la balance vers l’activation) NK. Cancer epithélial = carcinome. ULBP : UL16-binding protein. MIC : MHC I polypeptide related.")
49
V-Explications moléculaires de la réponse NK
- 3 - Réponse antivirale (EBV, CMV, Influenza…) : - Ligands de Récepteurs activateurs induits par infections virales (déséquilibre de la balance vers l’activation) - certains virus inhibent l’expression des CMH-I (herpesviridae, VIH…) NK Infection virale Ex : infection pulmonaire à influenza ULBP : UL16-binding protein MIC : MHC I polypeptide related
 : - Ligands de Récepteurs activateurs induits par infections virales (déséquilibre de la balance vers l’activation) - certains virus inhibent l’expression des CMH-I (herpesviridae, VIH…) NK. Infection virale. Ex : infection pulmonaire à influenza. ULBP : UL16-binding protein. MIC : MHC I polypeptide related.")
50
la perte d’expression du CMH-I peut entraîner une activation des NK
Le « Missing-self » : la perte d’expression du CMH-I peut entraîner une activation des NK La réponse cytotoxique par les T CD8 est CMH-I restreinte Les cellules tumorales / infectées peuvent down-réguler l’expression du CMH-I pour éviter d’être reconnues par les T8 cytotoxiques T8 NK NK T8 DOES NOT KILL KILLS KILLS cible cible cible NK = stratégie complémentaire aux T8 cytotoxiques pour lutter contre les cellules du non-soi ou soi altéré

Présentations similaires