Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Dystrophies Myotoniques
Isabelle Pénisson-Besnier Département de Neurologie CHU Angers

2
Maladie de Steinert (DM1)
Expansion anormale d’un triplet [CTG]n n = 5 à 37 chez sujets sains n = 50 à >2000 chez patients Située dans la région 3’ non codante du gène DMPK (DM protéine kinase) 2 gènes contigus: DMWD et SIX 5 L'expansion de l'ADN est détectable soit par la technique du Southern Blot, soit par polymerase chain reaction (PCR) qui donne une réponse plus rapide mais ne peut être employée que pour de courtes répétitions de triplets (5 à 300). 19q13.3
 2 gènes contigus: DMWD et SIX 5. L expansion de l ADN est détectable soit par la technique du Southern Blot, soit par polymerase chain reaction (PCR) qui donne une réponse plus rapide mais ne peut être employée que pour de courtes répétitions de triplets (5 à 300). 19q13.3.")
4
Genetic mapping of a second myotonic dystrophy locus Ranum LP, Rasmussen PF, Benzow KA, Koob MD, Day JW Department of Neurology and Institute of Human Genetics, University of Minnesota, Minneapolis 55455, USA Locus DM2 en 3q21 –> nouvelle nomenclature -> locus maladie de Steinert rebaptisé locus DM1 Nat Genet. 1998;19:196-8

5
Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF Liquori CL, Ricker K, Moseley ML, Jacobsen JF, Kress W, Naylor SL, Day JW, Ranum LP Institute of Human Genetics; MMC 206, 420 Delaware Street SE, University of Minnesota, Minneapolis, MN 55455, USA 1ère maladie rapportée à l'expansion d'un tétranucléotide Science 2001;293:864-7

6
Aspects génétiques Locus DM2 en 3q21
Expansion quadruplet CCTG dans intron 1 gène ZNF9 Allèles sains (TG)14-25 (TCTG)4-10 (CCTG)11-26 Dernière portion interrompue par des tétranucléotides Allèles porteurs de la mutation DM2 Nombre moyen répétitions CCGT: ( ) Perte des interruptions (prémutation) Instabilité somatique de l'expansion Variation intergénérationnelle Pas de phénomène d'anticipation (CCTG)n enfant < (CCTG)n parent (père ou mère) Corrélation positive entre taille expansion et âge début Protéine en doigt de zinc 9 (ZNF9) Taille expansion très >> à celle de DM1 Instabilité: nb de répétitions augmente avec le temps chez un individu donné (accroissement de 500 répétitions dans le sang d'un sujet prélevé à 3 ans d'intervalle)
14-25 (TCTG)4-10 (CCTG) Dernière portion interrompue par des tétranucléotides. Allèles porteurs de la mutation DM2. Nombre moyen répétitions CCGT: ( ) Perte des interruptions (prémutation) Instabilité somatique de l expansion. Variation intergénérationnelle. Pas de phénomène d anticipation. (CCTG)n enfant < (CCTG)n parent (père ou mère) Corrélation positive entre taille expansion et âge début. Protéine en doigt de zinc 9 (ZNF9) Taille expansion très >> à celle de DM1. Instabilité: nb de répétitions augmente avec le temps chez un individu donné (accroissement de 500 répétitions dans le sang d un sujet prélevé à 3 ans d intervalle)")
7
Epidémiologie DM2 379 individus issus de 133 familles (Day et al., 2003) Majorité des familles originaire du nord Europe (Allemagne, Pologne) Haplotype commun dans 17 familles DM2 européennes d'origine géographique distincte (Bachinski et al., 2003) Quelques familles hors Europe Prévalence minimale DM2 estimée à 1/ Prévalence DM1 = 5/
 Quelques familles hors Europe. Prévalence minimale DM2 estimée à 1/ Prévalence DM1 = 5/")
8
DM2 Age au début: entre 30 et 60 ans Symptômes d'appel
Pas de forme congénitale +++ ni infantile Symptômes d'appel Faiblesse musculaire Douleurs musculaires Myotonie Démasqués ou aggravés par hypothyroïdie (Sansone et al., 2000)
")
9
Manifestations musculaires
Douleurs Présentes dans ~ 50% cas (63% après 50 ans) Episodiques, fluctuantes, sans lien avec l'exercice, prédominent au repos Siègent préférentiellement aux membres inférieurs Diffuses ou pseudo-radiculaires Brûlures ou tiraillements Faiblesse Fléchisseurs du cou, abdominaux (précoce) Muscles proximaux des membres inférieurs (après 50 ans) Atteinte faciale, ptosis, dysarthrie: minimes ou absents Hypertrophie mollets. Amyotrophie dans < 10% cas Myotonie clinique Inconstante +++ (< 50% à 75% cas selon séries) Variable dans le temps Asymétrique et focale Exacerbée pendant la grossesse (Newman et al., 1999)
 Episodiques, fluctuantes, sans lien avec l exercice, prédominent au repos. Siègent préférentiellement aux membres inférieurs. Diffuses ou pseudo-radiculaires. Brûlures ou tiraillements. Faiblesse. Fléchisseurs du cou, abdominaux (précoce) Muscles proximaux des membres inférieurs (après 50 ans) Atteinte faciale, ptosis, dysarthrie: minimes ou absents. Hypertrophie mollets. Amyotrophie dans < 10% cas. Myotonie clinique. Inconstante +++ (< 50% à 75% cas selon séries) Variable dans le temps. Asymétrique et focale. Exacerbée pendant la grossesse (Newman et al., 1999)")
10
Autres manifestations (1)
Cataracte Présente dans environ 50% cas avant 60 ans Précoce (2/10 patients avant 20 ans) Sous-capsulaire postérieure, opacités multicolores irisées Calvitie Présente chez 1/3 des hommes entre 21 et 34 ans Atteinte cardiaque Moins fréquente et moins sévère que dans DM1 Troubles de conduction (BAVI, BB) sur ECG chez 20% patients Rares cas de décès par troubles du rythme, cardiomyopathie (7%) Hyperhydrose 20 à 30% des patients (Day et al., 2003)
 Sous-capsulaire postérieure, opacités multicolores irisées. Calvitie. Présente chez 1/3 des hommes entre 21 et 34 ans. Atteinte cardiaque. Moins fréquente et moins sévère que dans DM1. Troubles de conduction (BAVI, BB) sur ECG chez 20% patients. Rares cas de décès par troubles du rythme, cardiomyopathie (7%) Hyperhydrose. 20 à 30% des patients (Day et al., 2003)")
11
Autres manifestations (2)
Manifestations endocriniennes Troubles du métabolisme glucidique avec hyperinsulinisme Diabète (20% cas) HGPO anormale (75% cas) Hypogonadisme hypergonadotrope (chez 2/3 des hommes explorés) Elévation du taux de GGT Diminution du taux sérique des IgG et IgM Atteinte du système nerveux central Pas de cas de DM2 avec retard mental +++ Profil cognitif et comportemental particulier: altération des fonctions exécutives, traits de personnalité évitants (Meola et al, 2003) IRM 3-D: atrophie cérébrale (Kassubek et al., 2003) TEP: hypoperfusion cérébrale régions frontales et pariéto-occipitales Quelques cas avec anomalies de la substance blanche cérébrale
 HGPO anormale (75% cas) Hypogonadisme hypergonadotrope (chez 2/3 des hommes explorés) Elévation du taux de GGT. Diminution du taux sérique des IgG et IgM. Atteinte du système nerveux central. Pas de cas de DM2 avec retard mental +++ Profil cognitif et comportemental particulier: altération des fonctions exécutives, traits de personnalité évitants (Meola et al, 2003) IRM 3-D: atrophie cérébrale (Kassubek et al., 2003) TEP: hypoperfusion cérébrale régions frontales et pariéto-occipitales. Quelques cas avec anomalies de la substance blanche cérébrale.")
12
Meola et al., Neuromusc Disord 2003
Mastaglia et al., JNNP 1998 99mTc-ECD SPECT SPM group analysis in five patients with 3q-linked PROMM/DM-2. In red the brain regions with cerebral blood flow reductions Meola et al., Neuromusc Disord 2003

13
Examens paracliniques
Taux sérique de CK Normal ou peu élevé 1 cas d'hyper-CKémie isolée (Merlini et al., 2005) Electromyogramme Salves myotoniques inconstantes (Ricker, 1999) Diversité des activités EMG de repos (Ricker, 1999) Test d'effort bref pour distinguer DM1 et DM2 (Sander et al., 2000) Dosages sanguins (glycémie, GGT, immunoglobulines, testostérone,..) Biopsie musculaire Diagnostic génétique moléculaire
 Electromyogramme. Salves myotoniques inconstantes (Ricker, 1999) Diversité des activités EMG de repos (Ricker, 1999) Test d effort bref pour distinguer DM1 et DM2 (Sander et al., 2000) Dosages sanguins (glycémie, GGT, immunoglobulines, testostérone,..) Biopsie musculaire. Diagnostic génétique moléculaire.")
14
Results of the 10 s (n=7), 30 s (n=5), and 60 s (n=5) exercise test of abductor pollicis brevis (APB) in proximal myotonic myopathy (PROMM). Time (min) is plotted on the X axis, with exercise cessation occurring at 0 min. The Y axis represents the compound muscle action potential (CMAP) amplitude as a percentage of the pre-exercise baseline CMAP amplitude. The results are normal, with a maximum post-exercise CMAP amplitude decline of 8%. Results of a 10 and a 30 s exercise test of abductor pollicis brevis (APB) in a patient with myotonic dystrophy (MD). Time (min) is plotted on the X axis, with exercise cessation occurring at 0 min. The Y axis represents the compound muscle action potential (CMAP) amplitude as a percentage of the pre-exercise baseline CMAP amplitude. There are significant immediate post-exercise CMAP amplitude declines of 49% (10 s exercise test) and 31% (30 s exercise test). Test d'effort bref (10 secondes): amplitude du potentiel moteur franchement réduite dans DM1, stable dans DM2 Sander et al., Clin Neurophysiol 2000
 in a patient with myotonic dystrophy (MD). Time (min) is plotted on the X axis, with exercise cessation occurring at 0 min. The Y axis represents the compound muscle action potential (CMAP) amplitude as a percentage of the pre-exercise baseline CMAP amplitude. There are significant immediate post-exercise CMAP amplitude declines of 49% (10 s exercise test) and 31% (30 s exercise test). Test d effort bref (10 secondes): amplitude du potentiel moteur franchement réduite dans DM1, stable dans DM2. Sander et al., Clin Neurophysiol")
16
DM1 DM2 DM2 DM2 G. Bassez A B C D ATPase 9.4 ATPase 9.4 MHC slow
Muscle deltoïde. L’étude histoenzymologique (ATPase pH 9,4) montre des fibres de type 2 (foncées) de taille normale dans la DM1 (A), fréquemment atrophiques (indiquées par des flèches) dans la DM2 (B). Dans la DM2 (C,D), l’immunomarquage sur coupes sériées avec un anticorps dirigé contre l'isoforme lente (spécifique des fibres de type 1) (C), et rapide (identifiant les fibres de type 2) (D) de la chaîne lourde de la myosine, révèle un sous-groupe de fibres rapides de calibre extrêmement réduit ( D, flèches). MHC slow MHC fast G. Bassez
 montre des fibres de type 2 (foncées) de taille normale dans la DM1 (A), fréquemment atrophiques (indiquées par des flèches) dans la DM2 (B). Dans la DM2 (C,D), l’immunomarquage sur coupes sériées avec un anticorps dirigé contre l isoforme lente (spécifique des fibres de type 1) (C), et rapide (identifiant les fibres de type 2) (D) de la chaîne lourde de la myosine, révèle un sous-groupe de fibres rapides de calibre extrêmement réduit ( D, flèches). MHC slow. MHC fast. G. Bassez.")
17
Critères diagnostiques DM2
Faiblesse musculaire Myotonie à l'EMG Cataracte sous-capsulaire polychrome < 50 ans Hérédité autosomique dominante Nombre normal de triplets CTG pour le gène DMPK Fibres de type 2 très atrophiques, sacs nucléaires

18
Diagnostic génétique moléculaire DM2
Difficile +++ Southern blot conventionnel : expansion non détectée dans 20% cas prouvés DM2 PCR classique Exclut DM2 chez individus ayant 2 allèles normaux amplifiables Ne différencie pas individus sains avec 2 allèles de même taille (5 à 15% population générale), des individus atteints Mise au point de plusieurs étapes complémentaires Molecular diagnosis of DM2. (A) PCR analysis of the CL3N58 marker. The genotype of each individual is shown in base pairs. Alleles too large to amplify by PCR, which are referred to as "blank alleles," are indicated by a "B" and make the segregation of the markers appear non-Mendelian. (B) Expansion detection by genomic Southern analysis. DM2 Southern analysis of genomic DNA from control (N), affected individuals with a detectable (A) and nondetectable (A*) expanded allele is shown (Lanes 1–7). In contrast to DM2, an SCA8 Southern (Lane 8) shows equally intense signals for the normal and expanded alleles. (C) Schematic diagram of the PCR-based RA. The straight arrow represents the flanking primer CL3N58-D R. Tailed arrows represent the JJP4CAGG primer. A third primer (JJP3, not shown), used to make the PCR reaction more robust, has the same sequence as the hanging tail of JJP4CAGG. The primer used to probe Southern blots of the PCR products is CL3N58-E R. (D) Repeat assay results. RA results for affected individuals with expansions that were detected (A) or not detected (A**) by genomic Southern analysis and are shown in Lanes 1 to 5 and 8. Negative results from unaffected controls (N) are shown in Lanes 6,7,9, and 10. Day et al., Neurology 2003
, des individus atteints. Mise au point de plusieurs étapes complémentaires. Molecular diagnosis of DM2. (A) PCR analysis of the CL3N58 marker. The genotype of each individual is shown in base pairs. Alleles too large to amplify by PCR, which are referred to as blank alleles, are indicated by a B and make the segregation of the markers appear non-Mendelian. (B) Expansion detection by genomic Southern analysis. DM2 Southern analysis of genomic DNA from control (N), affected individuals with a detectable (A) and nondetectable (A*) expanded allele is shown (Lanes 1–7). In contrast to DM2, an SCA8 Southern (Lane 8) shows equally intense signals for the normal and expanded alleles. (C) Schematic diagram of the PCR-based RA. The straight arrow represents the flanking primer CL3N58-D R. Tailed arrows represent the JJP4CAGG primer. A third primer (JJP3, not shown), used to make the PCR reaction more robust, has the same sequence as the hanging tail of JJP4CAGG. The primer used to probe Southern blots of the PCR products is CL3N58-E R. (D) Repeat assay results. RA results for affected individuals with expansions that were detected (A) or not detected (A**) by genomic Southern analysis and are shown in Lanes 1 to 5 and 8. Negative results from unaffected controls (N) are shown in Lanes 6,7,9, and 10. Day et al., Neurology")
19
Day & Ranum, Neuromusc Disord 2005
Pathogenic model of DM1 and DM2. The model of RNA pathogenesis in DM1 and DM2 is due to the untranslated expansions in each disease. Both expansions are transcribed; the DMPK mRNA containing the CUG expansion is incorporated into the ribonuclear inclusions; the CCUG expansion from the DM2 transcript is incorporated into ribonuclear inclusions, though it remains unclear whether any other elements of the ZNF9 transcript are also contained within the inclusions. Muscleblind protein (MBNL) binds to the ribonuclear inclusions; CUG-BP is increased by unclear mechanisms. Decreased MBNL and increased CUG-BP activity alter splicing of transcripts involved in DM pathogenesis, e.g. transcripts encoding the chloride channel and insulin receptor. Although the genes responsible for some clinical features have not yet been identified (e.g. testicular failure and hypogammaglobulinemia), the occurrence of these abnormalities in both DM1 and DM2 indicates that they are likely to be caused by the toxic effects of repeat expansions in RNA, possibly involving the resultant decrease in MBNL and increase in CUG-BP. Day & Ranum, Neuromusc Disord 2005
 binds to the ribonuclear inclusions; CUG-BP is increased by unclear mechanisms. Decreased MBNL and increased CUG-BP activity alter splicing of transcripts involved in DM pathogenesis, e.g. transcripts encoding the chloride channel and insulin receptor. Although the genes responsible for some clinical features have not yet been identified (e.g. testicular failure and hypogammaglobulinemia), the occurrence of these abnormalities in both DM1 and DM2 indicates that they are likely to be caused by the toxic effects of repeat expansions in RNA, possibly involving the resultant decrease in MBNL and increase in CUG-BP. Day & Ranum, Neuromusc Disord")
20
Pathogénie

21
(CAGG)8 sense oligonucleotide
DM2 DM2 (CCTG)8 sense oligonucleotide (CAGG)8 antisense oligonucleotide Contrôle DM1 Visualization of the DM2 (CCTG)n expansion mutation by CISH on muscle sections. Specific DAB label is seen as dark brown deposits within the myonuclei. A single-spot signal representing the genomic DM2 mutation is obtained with the (CCTG)8 sense oligonucleotide (a, DM2 patient frozen muscle), while the ribonuclear inclusions containing accumulated mutant RNAs are detected with the (CAGG)8 antisense oligonucleotide (b, same DM2 patient as in a, frozen muscle). Gill's hematoxylin was used as a counterstain. A DM1 control muscle hybridized with the (CAGG)8 antisense probe showing no signal (c). The (CCTG)8 sense probe hybridized on frozen muscle of a healthy control gives no signal (d), (CAGG)8 sense oligonucleotide (CCTG)8 sense oligonucleotide Sallinen et al., Neuromusc Disord 2004
8 sense oligonucleotide. (CAGG)8 antisense oligonucleotide. Contrôle. DM1. Visualization of the DM2 (CCTG)n expansion mutation by CISH on muscle sections. Specific DAB label is seen as dark brown deposits within the myonuclei. A single-spot signal representing the genomic DM2 mutation is obtained with the (CCTG)8 sense oligonucleotide (a, DM2 patient frozen muscle), while the ribonuclear inclusions containing accumulated mutant RNAs are detected with the (CAGG)8 antisense oligonucleotide (b, same DM2 patient as in a, frozen muscle). Gill s hematoxylin was used as a counterstain. A DM1 control muscle hybridized with the (CAGG)8 antisense probe showing no signal (c). The (CCTG)8 sense probe hybridized on frozen muscle of a healthy control gives no signal (d), (CAGG)8 sense oligonucleotide. (CCTG)8 sense oligonucleotide. Sallinen et al., Neuromusc Disord")
22
Syndromes Myotoniques
Affections génétiques Dystrophies myotoniques (DM1, DM2, ..) Maladies des canaux ioniques musculaires Myotonie congénitale (canal chlore) Paramyotonie et paralysie périodique hyperkaliémique (canal sodium) Syndrome d'Andersen (canal potassium) Syndrome de Schwartz-Jampel (perlecan) Causes acquises médicamenteuses (fibrates et statines, chloroquine, colchicine)
 Maladies des canaux ioniques musculaires. Myotonie congénitale (canal chlore) Paramyotonie et paralysie périodique hyperkaliémique (canal sodium) Syndrome d Andersen (canal potassium) Syndrome de Schwartz-Jampel (perlecan) Causes acquises médicamenteuses (fibrates et statines, chloroquine, colchicine)")
23
Adolescence à adulte tardif
DM 1 (Steinert) DM 2 (PROMM) Epidémiologie Quasi-universelle Surtout en Europe Age de début Tout âge Adolescence à adulte tardif Anticipation + - Forme congénitale Faiblesse Faciale Proximale Distale ++ +/- Myalgies Hypertrophie mollets Cataracte Calvitie Troubles cardiaques Hypogonadisme Hyperglycémie Hypersomnie diurne Myotonie à l'EMG Retard mental Locus 19q13.3 3q21 Gène muté DMPK ZNF9 Expansion Triplets CTG Quadruplets CCTG Taille expansion 50 à 4 000 75 à (moyenne 5 000)
 DM 2 (PROMM) Epidémiologie. Quasi-universelle. Surtout en Europe. Age de début. Tout âge. Adolescence à adulte tardif. Anticipation. + - Forme congénitale. Faiblesse. Faciale. Proximale. Distale. ++ +/- Myalgies. Hypertrophie mollets. Cataracte. Calvitie. Troubles cardiaques. Hypogonadisme. Hyperglycémie. Hypersomnie diurne. Myotonie à l EMG. Retard mental. Locus. 19q q21. Gène muté. DMPK. ZNF9. Expansion. Triplets CTG. Quadruplets CCTG. Taille expansion. 50 à à (moyenne 5 000)")
24
Famille MA…. 13.3 kb 14 kb René (1922-90) Lucien (1923-96) Gérard
dcd à 40 ans (K) Michel Cataracte op.< 50 ans André 1951 J. Marc 1952 Isabelle (1957) Véronique (1961) Patrick ( ) Brigitte 13.3 kb 14 kb
 Michel. Cataracte. op.< 50 ans. André J. Marc Isabelle. (1957) Véronique. (1961) Patrick. ( ) Brigitte kb. 14 kb.")
25
Cas 1 (BM) Axe Membres supérieurs Membres inférieurs Walton score 2/4-
Fléchisseurs/Extenseurs cou Abdominaux 2/4- 2 Membres supérieurs Deltoïde Biceps/Triceps Palmaires/Radiaux Fléchisseurs/Extenseurs doigts 3+ 4/3+ 3+/3+ Membres inférieurs Psoas Grands fessiers/Moyens fessiers Quadriceps/Ischio-jambiers Triceps/Tibial antérieur 4+/4- 4+/4+ 5/3+ Walton score

26
Scanner musculaire Brigitte MA…. (31-05-1955)
")
27
Brigitte MA…. ( ) EMG: salves myotoniques (TA), décharges répétitives complexes CK normales Thrombopénie, anémie, hématies en larmes, IgG monoclonale lambda Glycémie, TSH normales Biologie hépatique N Capacité vitale, GDS N ECG: HBASG, Holter: qq ESV, échocardiographie N

28
Famille DES…. Pierre (1925) Jean (1929-1987) Marcel M. Louise
( ) Claude ( ) Francine 1941 Isabelle (1957) Véronique (1961) Gilbert 1950 Henri ( )
 Claude. ( ) Francine Isabelle. (1957) Véronique. (1961) Gilbert Henri. ( )")
29
Cas 1 (BM) Cas 2 (FD) Axe Membres supérieurs Membres inférieurs
Fléchisseurs/Extenseurs cou Abdominaux 2/4- 2 2/3+ 2- Membres supérieurs Deltoïde Biceps/Triceps Palmaires/Radiaux Fléchisseurs/Extenseurs doigts 3+ 4/3+ 3+/3+ 3+/4- 4/4 Membres inférieurs Psoas Grands fessiers/Moyens fessiers Quadriceps/Ischio-jambiers Triceps/Tibial antérieur 4+/4- 4+/4+ 5/3+ 3+/3- 4-/3 5/4- Walton score

30
Francine DES… ( ) EMG: pas de myotonie, tracés de type myopathique CPK 1 à 2.1 N GGT 6.7 N, ASAT 1.1 N, ALAT 1.7 N, PA 1.1 N Glycémie, TSH N Diminution taux sérique IgG et IgM ECG, Holter, échocardiographie N CV et gazométrie N Gène DM1: pas de mutation Gène DM2 : > 15 Kb

31
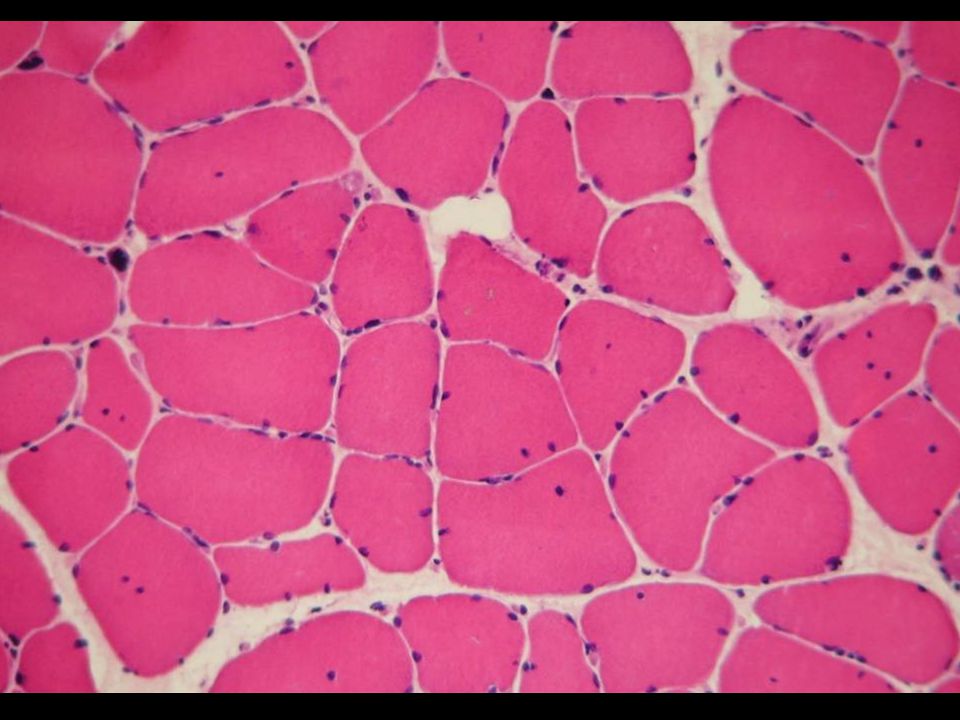
Variation calibre fibres +++ + Internalisations nucléaires ++
(muscle deltoïde) LM BM FD Variation calibre fibres +++ + Internalisations nucléaires ++ Sacs nucléaires Nécrose-régénération - Anomalies structure Ragged red fibers Moth-eaten fibers Fibres lobulées Prédominance fibres type 1 Fibrose endomysiale Infiltration adipeuse Autres Micropérivascularite périmysiale
 LM. BM. FD. Variation calibre fibres Internalisations nucléaires. ++ Sacs nucléaires. Nécrose-régénération. - Anomalies structure. Ragged red fibers. Moth-eaten fibers. Fibres lobulées. Prédominance fibres type 1. Fibrose endomysiale. Infiltration adipeuse. Autres. Micropérivascularite. périmysiale.")
32
Conclusions Savoir évoquer DM2 devant
Déficit myopathique d'installation tardive, à prédominance axiale et proximale Volontiers associé à des douleurs A fortiori si patient porteur d'une cataracte précoce Intérêt de l'EMG et de la biopsie musculaire Etude prospective (Udd, 2005) chez 52 patients avec myopathie de cause indéterminée 16 patients avec mutation DM2 (30%) Dont 3 avec phénotype DM2, sans histoire familiale DM3 (locus 15q-24) associée à une démence fronto-temporale (Le Ber et al, 2004)
 chez 52 patients avec myopathie de cause indéterminée. 16 patients avec mutation DM2 (30%) Dont 3 avec phénotype DM2, sans histoire familiale. DM3 (locus 15q-24) associée à une démence fronto-temporale (Le Ber et al, 2004)")
Présentations similaires




>")





>")
 Pr E. Tournier-Lasserve>")



 Pr E. Tournier-Lasserve>")




