Télécharger la présentation
1
Les empreintes génomiques
Certains gènes s’expriment à partir du chromosome paternel ou maternel mais pas les deux Quels sont les mécanismes derrières ce choix? Quels sont les conséquences niveau santé Quel est l’intérêt pour des cellules diploïdes d’utiliser un seul gène quand 2 sont présents

2
Transfert des pronoyau

3
Disomy et développement

4
Les gènes avec des empreintes sont méthylés ( circles rouges) dans les gamètes. La méthylation est maintenue dans les cellules somatiques. Par contre dans les cellules germinales il faut que les empreintes soit effacés et ensuite rétablis.

5
Un gène peut avoir une empreinte dans un ou plusieurs tissus

6
Dopa carboxylase (Ddc): 2 transcripts produits à partir des allèles maternels et paternels dans tous les tissus sauf le coeur Grb10:Paternel allèle exprimé dans le cerveaucomportement social Maternel allèle exprimé partoutmétabolism de glucose et homeostase

7
La demethylation au debut du développement

8
Imprinting Models

9
Empreintes sur les gènes H19 et Igf2 L’ADN maternel n’est pas méthylé et le CTCF peut se fixer. Le CTCF empêche l’enhancer d’activer l’expression du gène Igf2. La méthylation sur le chromosome paternel empêche le CTCF de se fixer et l’enhancer peut activer l’expression du gène Igf2

10
Les ARN non-codants sont souvent associés avec les empreintes génétiques

11
Informations générales sur les empreintes….
-dans les régions avec des empreintes il y a souvent plusieurs gènes sous le contrôle d’un IC (imprinting centre) -les empreintes ne sont pas toujours conservés entre la souris et l’homme -les gènes avec une empreinte ( pas exprimé) sont répliqués tard dans le cycle cellulaire ( marquage avec BrdU) -LOI ( loss of imprinting) est souvent associé avec le développement des cancers ( Igf2 et Wilms tumour, BWS) -les gènes avec des empreintes n’ont pas une distribution uniforme: 50% se trouve sur le chromosome 7 chez la souris -le chromosome X dans le trophoblast montre une empreinte
 -les empreintes ne sont pas toujours conservés entre la souris et l’homme. -les gènes avec une empreinte ( pas exprimé) sont répliqués tard dans le cycle cellulaire ( marquage avec BrdU) -LOI ( loss of imprinting) est souvent associé avec le développement des cancers ( Igf2 et Wilms tumour, BWS) -les gènes avec des empreintes n’ont pas une distribution uniforme: 50% se trouve sur le chromosome 7 chez la souris. -le chromosome X dans le trophoblast montre une empreinte.")
12
La transcription de l’ARN Kcnq1ot1 mène à la production d’un domaine où il n’y a pas de transcription. RNA-FISH (Kcnq1ot1-vert) DNA-FISH-rouge (Igf2-sans empreinte, Ascl2,empreinte placenta, Cdkn1c empreinte placenta et embryon)
 DNA-FISH-rouge (Igf2-sans empreinte, Ascl2,empreinte placenta, Cdkn1c empreinte placenta et embryon).")
13
Rôle des ARN non-codant dans la répression de la transcription
Délétion du promoteur du gène Kcnq1ot1: expression de plusieurs gènes normalement sous l’empreinte et non-exprimés Modification de l’état de méthylation dans les regions DMR: RIP (RNA Immunoprecipitation) avec anticorps contre Dnmt1 Interaction soit direct ( peut-être indirect?) entre l’ARN non-codant et la méthylase. La présence du transcrit guide l’enzyme sur les sites qu’il faut méthylé ….Kcnq1ot1 peut aussi recruter les protéines qui inhibent la transcription ( polycomb, histones modifiées)
 avec anticorps contre Dnmt1 Interaction soit direct ( peut-être indirect ) entre l’ARN non-codant et la méthylase. La présence du transcrit guide l’enzyme sur les sites qu’il faut méthylé ….Kcnq1ot1 peut aussi recruter les protéines qui inhibent la transcription ( polycomb, histones modifiées)")
14
Expression et la réplication des gènes avec les empreintes génomiques
Les gènes avec des empreintes sont répliqués tard dans le cycle cellulaire Les empreintes sont établis pendant la gametogenèse

15
Réplication des gènes avec des empreintes
-Culture de cellules avec la mitose synchronisé -marquage avec BrdU -analyse d’un gène avec une empreinte qui est polymorphique entre mâle et femelle -analyse des échantillons pendant le cycle cellulaire par Southern blot et les anti-corps contre BrdU -l’allèle sans l’empreinte ( exprimé) est toujours répliqué avant l’autre Juste avant la méiose les empreintes sont modifiés et tous les gamètes portent le même empreinte
 est toujours répliqué avant l’autre. Juste avant la méiose les empreintes sont modifiés et tous les gamètes portent le même empreinte.")
16
Beckwith-Wiedemann Syndrome 2 loci sur le chromosome 11 CDKN1C: inhibiteur d’un kinase qui freine la proliferation cellulaire IGF2: Insulin Like Growth factor 2 Absence del’expression de CDKN1C et trop d’expression de IGF2syndrome a

17
Beckwith-Wiedemann Syndrome
-Phénotype variable -Souvent enfants obèse -plus de cancer ( Wilm’s Tumour, Rhabdomyosarcoma) -Plus fréquente chez les jumelle monozygotique -anomalies avec les oreilles et la langue -85% des cas sont sporadiques -20% meurt avant 12 mois
 -Plus fréquente chez les jumelle monozygotique. -anomalies avec les oreilles et la langue. -85% des cas sont sporadiques. -20% meurt avant 12 mois.")
18
Angelman Prader Willi

19
Les syndromes de Prader-Willi et Angelman

20
Mechanisms of imprinting of the Prader–Willi/Angelman region
Imprinted gene expression and epigenetic marks in human chromosome region 15q11–q13. Color coding of boxes indicates whether genes show paternal‐only expression, paternal > maternal expression, maternal > paternal expression, or equal expression of paternal and maternal alleles. PWRN1 and C15Orf2 show monoallelic expression in human fetal brain, but parental origin of the monoallelic expression has not been determined; because these genes are within a cluster of genes with paternal‐only expression, these genes are color coded as paternal > maternal. Parent‐specific epigenetic modifications are shown as symbols on vertical black lines. Figure is not drawn to scale. © This slide is made available for non-commercial use only. Please note that permission may be required for re-use of images in which the copyright is owned by a third party. American Journal of Medical Genetics Part A Volume 146A, Issue 16, pages , 14 JUL 2008 DOI: /ajmg.a

21
Mechanisms of imprinting of the Prader–Willi/Angelman region
Model for control of gene expression in 15q11–q13 by the PWS/AS‐IC. On the paternal copy of chromosome 15, the PWS‐SRO is unmethylated and active. This unmethylated PWS‐SRO acts by unknown mechanisms to activate transcription of MKRN3, MAGEL2, and NDN, and it causes brain‐specific silencing of UBE3A by mechanisms that may include serving as promoter for an antisense transcript. On the maternal copy of chromosome 15, the PWS‐SRO is methylated and inactive. In the absence of an active PWS‐SRO, MKRN, MAGEL2, NDN, and SNRPN are silenced and UBE3A is transcriptionally active in brain. In patients with PWS‐SRO deletions, the consequences for gene expression are the same as on a maternal chromosome with PWS‐SRO methylation. In patients with AS‐SRO deletions who have intact PWS‐SRO, the PWS‐SRO is not methylated after maternal transmission, so that the consequences for gene expression are the same as on a normal paternal chromosome. In patients with deletions encompassing both AS‐SRO and PWS‐SRO (not shown), the lack of an active PWS‐SRO leads to a maternal pattern of gene expression. © This slide is made available for non-commercial use only. Please note that permission may be required for re-use of images in which the copyright is owned by a third party. American Journal of Medical Genetics Part A Volume 146A, Issue 16, pages , 14 JUL 2008 DOI: /ajmg.a
, the lack of an active PWS‐SRO leads to a maternal pattern of gene expression. © This slide is made available for non-commercial use only. Please note that permission may be required for re-use of images in which the copyright is owned by a third party. American Journal of Medical Genetics Part A Volume 146A, Issue 16, pages , 14 JUL 2008 DOI: /ajmg.a")
22
Prader-Willi et Angelman Syndrome

23
Trisomy Rescue

24
PWS:Une maladie complexe: critères du diagnostic clinique (Cassidy 2001)
Critères majeurs (>60%) Hypotonie néonatale sévère Retard global du développement Hyperphagie qui conduit à une obésité Problèmes de comportement (troubles psychiatriques) Hypogonadisme Déficits cognitifs Défauts d’articulation Grattage Mains et pieds de petite taille Critères mineurs (<50%) Anormalités des yeux Scoliose, ostéoporose Température instable Insensibilité à la douleur Apnées du sommeil dues à des problèmes de respiration Caractéristiques faciales Habileté à réaliser des puzzles
 Hypotonie néonatale sévère. Retard global du développement. Hyperphagie qui conduit à une obésité. Problèmes de comportement (troubles psychiatriques) Hypogonadisme. Déficits cognitifs. Défauts d’articulation. Grattage. Mains et pieds de petite taille. Critères mineurs (<50%) Anormalités des yeux. Scoliose, ostéoporose. Température instable. Insensibilité à la douleur. Apnées du sommeil dues à des problèmes de respiration. Caractéristiques faciales. Habileté à réaliser des puzzles.")
25
PWS:Une maladie évolutive
0 à 2 ans: hypotonie importante. Problèmes de succion et de déglutition 2 à 6 ans: histoire de l’hypotonie + retard du développement 6 à 12 ans: hyperphagie et problèmes de comportement 13 ans à adulte: fonctionnement intellectuel réduit ,hyperhagie, obésité, hypogonadisme comportement obsessif-compulsif ,crises de colères.

26
Les modifications génétiques chez les personnes atteints de PWS donnent les phénotypes différents
IQ range : Pour tous les Prader-Willi 20 – 100 ; mean = 50 – 70 Verbal IQ: mieux chez les malades avec uniparental disomy Capacité pour faire des puzzles: mieux chez les malades avec une déletion Mémoire pour les photos etc: mieux avec le uniparental disomy D’une manière générale leur progrès académique est moins bon par rapport à leurs capacités cognitifs

27
NECDIN is implicated in PWS
NECDIN is never expressed in Prader-Willi patients. NECDIN is strongly expressed in the Nervous System Necdin-deficient mice phenotype mimics PWS : - Partial early postnatal lethality (respiratory distress) - Growth developmental delay - Increased skin scraping activity - Improvement of spatial learning - Sensory-motor defects - Alteration of pain threshold Reduction of the number of GnRH and oxytocin neurons ……incomplete studies (obesity with high fat diet..) Mutant Wild-type P8 (Human Molecular Genetics 2000, Manuscript submitted and data not published)
 - Growth developmental delay. - Increased skin scraping activity. - Improvement of spatial learning. - Sensory-motor defects. - Alteration of pain threshold. Reduction of the number of GnRH and oxytocin neurons. ……incomplete studies (obesity with high fat diet..) Mutant. Wild-type. P8. (Human Molecular Genetics 2000, Manuscript submitted and data not published)")
28
Cellular function in sensory neurons
Increase of 41% apoptosis in Necdin deficient embryos +/+ +/- NF Tunel Brdu Tunel Loss of specified sensory neurons (at E13.5, P0) (Manuscript submitted)
 (Manuscript submitted)")
29
Test psychomotrice

30
Stochastic loss of silencing of the imprinted Ndn/NDN allele, in a mouse model and humans with prader-willi syndrome, has functional consequences. Rieusset A, Schaller F, Unmehopa U, Matarazzo V, Watrin F, Linke M, Georges B, Bischof J, Dijkstra F, Bloemsma M, Corby S, Michel FJ, Wevrick R, Zechner U, Swaab D, Dudley K, Bezin L, Muscatelli F. PLoS Genet. 2013;9(9):
:")
31
Angelman Syndrome People with Angelman syndrome love water and have a fascination for reflective surfaces, plastic, and balloons. They enjoy being in the company of others and watching TV, especially slapstick humour. With progression into adulthood the behaviour becomes quieter and concentration span increases

32
Angelman Syndrome Absence de l’expression du gène Ube3A: ubiquitin ligase essentiel pour envoyer les protéines vers le proteasome pour dégradation. Délai de développement important, QI très réduit. Peu de language, ataxia. Happy puppet: mouvement des bras comme un marionnette. Très souriante. Durée de vie normale; systèmes reproductives normales Incidence: 1/15000

33
Figure 1. In normal individuals, UBE3A is expressed from both the paternal (blue) and maternal (magenta) chromosomes 15 in most tissues Chamberlain, S. J. et al. J. Neurosci. 2010;30: Copyright ©2010 Society for Neuroscience

34
Figure 2. Genomic imprinting of chromosome 15q11-q13 and epigenetic silencing of UBE3A in neurons
Chamberlain, S. J. et al. J. Neurosci. 2010;30: Copyright ©2010 Society for Neuroscience

35
Comment fonctionne la protéine UBE3A
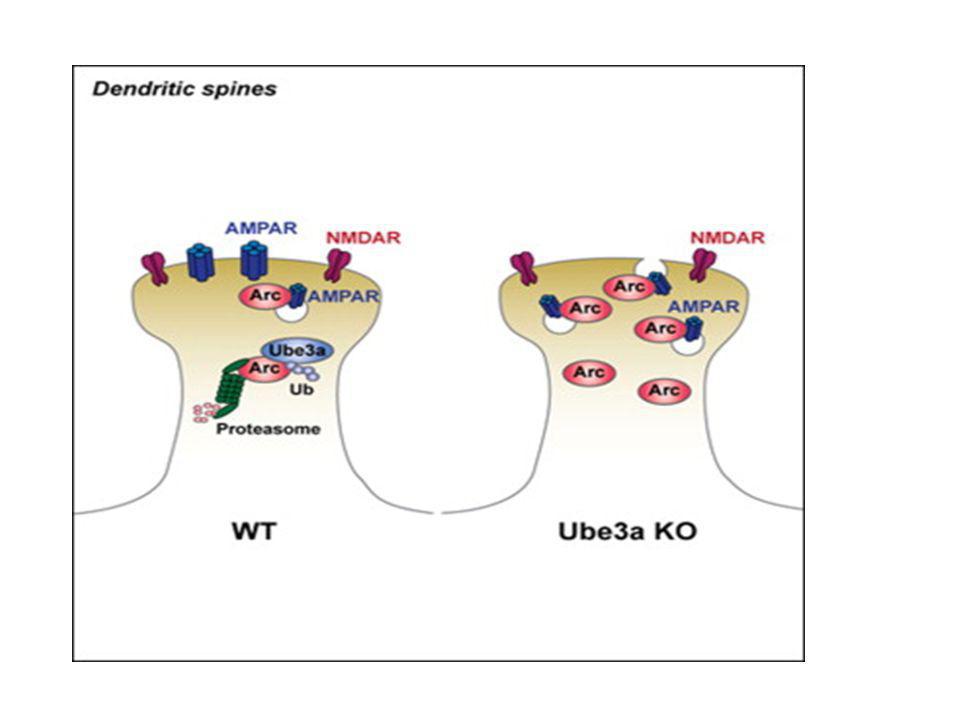
L’activité neuronal activation de la transcription du gène Ube3A: plus de transcription chez la souris dans un environnement ‘intéressant UBE3A important pour la dégradation d’une protéine ARC En présence de ARC le nombre de récepteurs AMPA est réduit aux synapses Les synapses avec moins de récepteurs APMA marchent moins bien pendant l’apprentisagesyndrome d’Angelman Cell

37
Pourquoi existent-elles les empreintes génomiques?
1. Compétition entre les mâles et les femelles. Une femelle peut avoir plusieurs partenaires et du coup des embryons avec des pères différents dans la même portée. Les empreintes femelles sont pour limiter la croissance des embryons pour que tous les embryons puissent survivre. Les empreintes mâles aident la croissance des embryons: chaque mâle veut que ses propres embryons survivent et pas forcement les autres. 2. Ovarian time bomb: le développement d’une ovocyte non-fécondé peut facilement donner un cancer. Pour éviter ce possibilité il faut inactiver les gènes dans le génome femelle qui augmente la croissance et activer les gènes qui freinent la croissance. Chez le mâle il faut inhiber l’expression des inhibiteurs pour permettre un développement normal. 3. Pour permettre une évolution plus rapide de l’allèle non-exprimé 4. Pour assurer une contribution mâle et femelle dans chaque embryon 5. L’expression des deux allèles est létale pour la cellule




>")

 Pr E. Tournier-Lasserve>")

 Pr E. Tournier-Lasserve>")












