Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Dr BENFETIMA S. Clinique pédiatrique du Mansourah
Les anémies hémolytiques de l’enfant Dr BENFETIMA S. Clinique pédiatrique du Mansourah

2
Definition Destruction anle du GR => ↓ durée de vie. mixte
mécanisme corpusculaire constitutionnelle Hémoglobine Enzyme Membrane Extra corpusculaire acquise Coombs positif Coombs négatif mixte

3
Interet : Fréquence des AH congénitales. Gravité.
Prise en charge très difficile. Prévention possible / Conseil génétique.

4
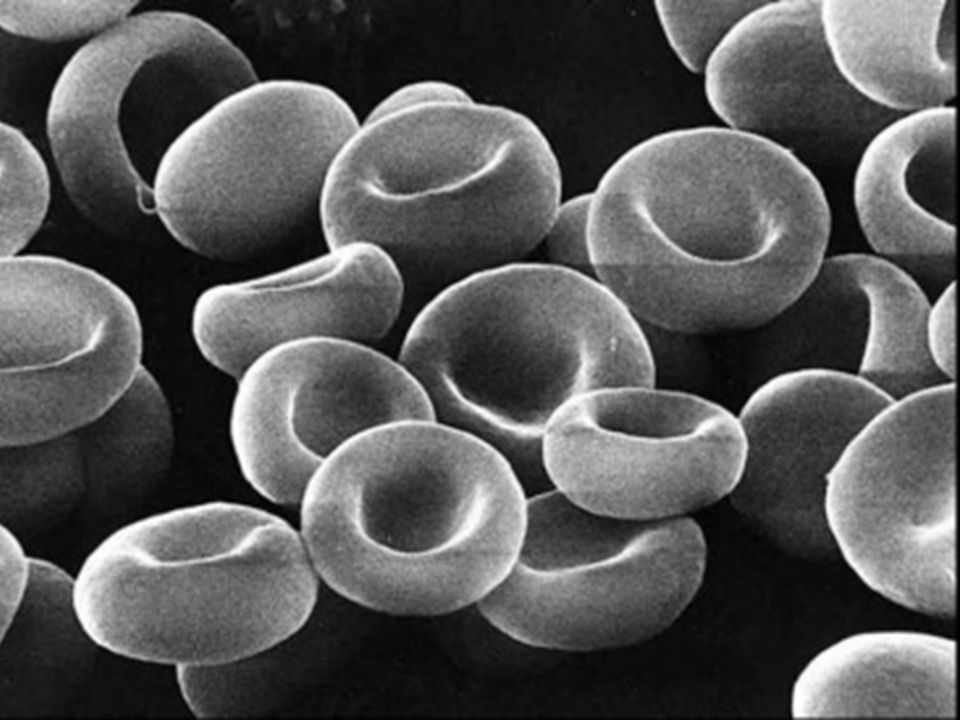
Le GR naît dans la moelle osseuse.
Vit 120 jours [ 1% par Jour ].

7
Hémolyse pathologique
Destruction prématurée des GR. Extravasculaire. Rarement intravasculaire. Avortement de l’érythropoeise.

9
Hyperactivité médullaire = réticulocytose + érythroblastose médullaire.
· compensation parfaite (sans anémie) = hémolyse compensée. · hémolyse > la production = anémie hémolytique décompensée.
 = hémolyse compensée. · hémolyse > la production = anémie hémolytique décompensée.")
10
Hemolyse aigue (intra-vasculaire):
Anémie aiguë + urines foncées. Ictère retardé. SPM inconstante. Anémie regenerative au bout de qlq h. Haptoglobinémie effondrée, Hbémie plasmatique et Hburie. Hyperbilirubinémie libre modérée. Dans les heures qui suivent : réticulocytose parfois hyperleucocytose. Thrombocytose. Erythroblastose medullaire.

11
Hemolyse chronique : Extravasculaire (ou intra tissulaire) :
triade : anémie + ictère + splénomégalie. Anémies régénératives(sauf thalassémie) BIL > 10 mg / L. Intra-vasculaire : IDEM + Haptoglobinémie effondrée + particularité : une hémosidérinurie.
 BIL > 10 mg / L. Intra-vasculaire : IDEM + Haptoglobinémie effondrée + particularité : une hémosidérinurie.")
12
Enquete etiologique : INTERROGATOIRE: EXAMENS BIOLOGIQUES:
RECHERCHE D’UN MECANISME CORPUSCULAIRE. RECHERCHE D’UN MECANISME EXTRA-CORPUSCULAIRE.

13
Efficacité des transfusions

14
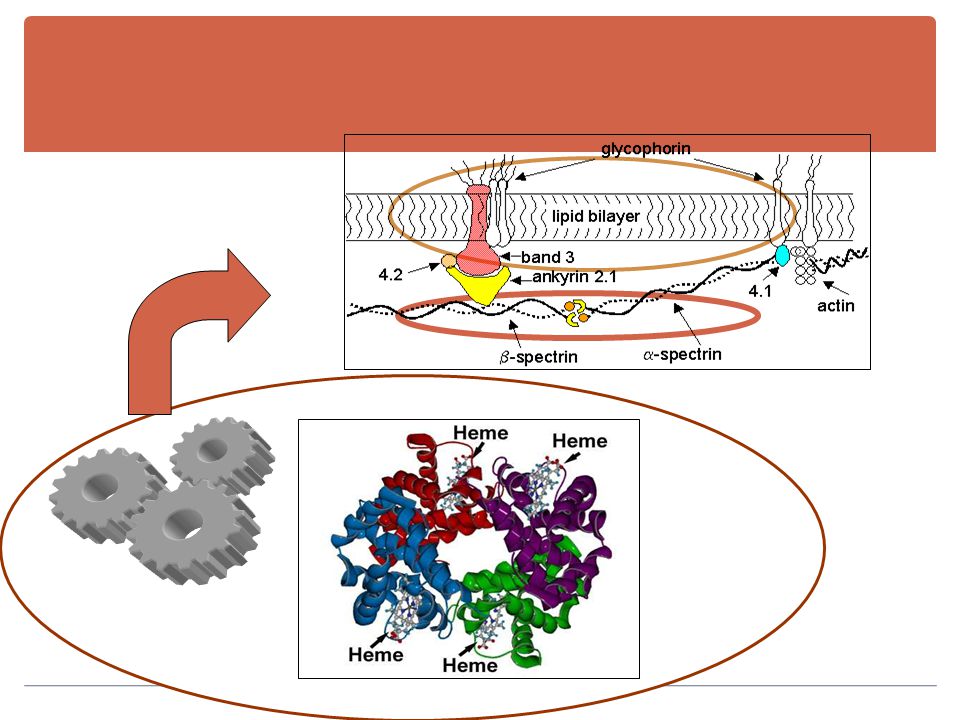
A.H. CORPUSCULAIRES : constitutionnelles
Anomalies de membrane . Hémoglobinopathies . Anomalies enzymatiques . A.H. EXTRA- CORPUSCULAIRES: acquises : Coombs [+] ou immunologiques. Coombs [ ̶ ]. A.H. MIXTES

15
AH constitutionnelles Anomalies de membrane
AH constitutionnelles Anomalies de membrane . Anomalies enzymatiques Hémoglobinopathies . .

16
Microsphérocytose héréditaire Maladie de Minkowski-Chauffard

18
Microsphérocytose héréditaire Maladie de Minkowski-Chauffard.
autosomale dominante (3/4). membrane: perd sa déformabilité, sa souplesse, se lyse facilement => Destruction intrasplénique +++. Hémolyse chronique avec poussées paroxystiques Enquête familiale : Histoire familiale. Cytologie : Présence de microsphérocytes. Test de fragilité osmotique. Test d’autohemolyse in vitro. Ektacytometrie, cytometrie en flux. Splénectomie.
. membrane: perd sa déformabilité, sa souplesse, se lyse facilement => Destruction intrasplénique +++. Hémolyse chronique avec poussées paroxystiques. Enquête familiale : Histoire familiale. Cytologie : Présence de microsphérocytes. Test de fragilité osmotique. Test d’autohemolyse in vitro. Ektacytometrie, cytometrie en flux. Splénectomie.")
19
Stomatocytose héréditaire ( hématies en bouche).
.")
20
Acanthocytose constitutionnelle

21
Elliptocytose familiale ( ovale).
.")
22
AH constitutionnelles Anomalies de membrane
AH constitutionnelles Anomalies de membrane . Anomalies enzymatiques Hémoglobinopathies . .

25
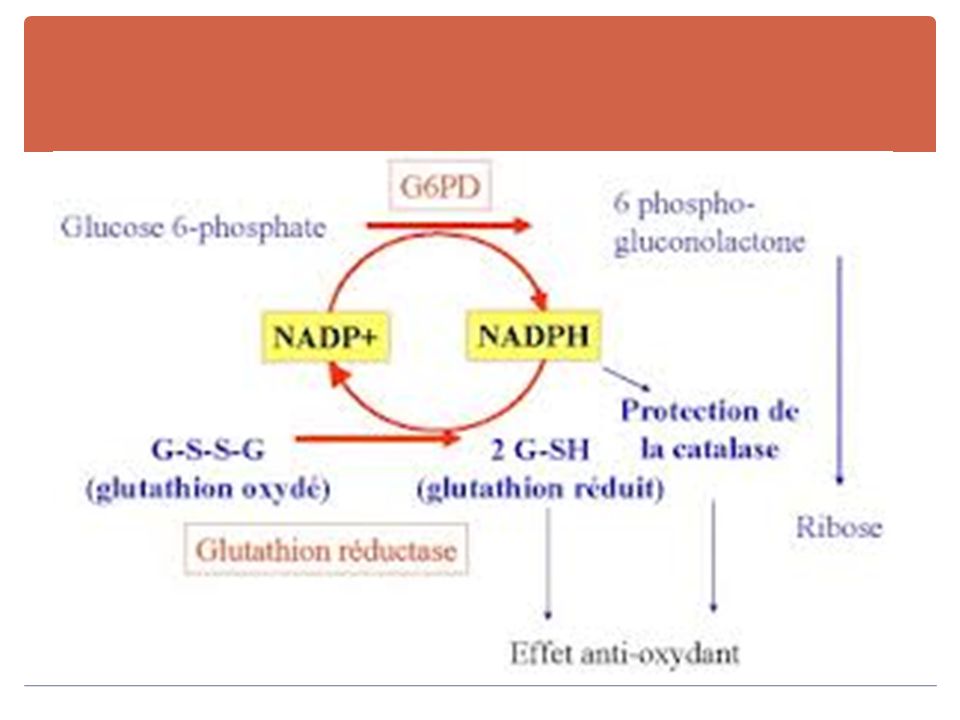
3. Déficits enzymatiques a. Déficit en G 6 PD
récessive liée au sexe. Quelques heures après la prise d'un agent déclenchant (médicaments, fèves, parfois infections), survient une crise brutale d'hémolyse IV Dosage du G6PD. Transfusion si anémie sévère. prévention : exclusion des fèves, certains médicaments dont la liste est remise au porteur du déficit. b. Déficits en pyruvate kinase et autres désordres de la glycolyse
, survient une crise brutale d hémolyse IV. Dosage du G6PD. Transfusion si anémie sévère. prévention : exclusion des fèves, certains médicaments dont la liste est remise au porteur du déficit. b. Déficits en pyruvate kinase et autres désordres de la glycolyse.")
26
AH constitutionnelles Anomalies de membrane. Anomalies enzymatiques
AH constitutionnelles Anomalies de membrane . Anomalies enzymatiques . Hémoglobinopathies .

27
2. Anomalie de l’Hb: Hémoglobine normale:
=> HbA1 [ alpha2, beta2 ] (97%) => HbA2 [ alpha2, delta2 ] (3%) => HbF [ alpha2, gamma2 ] (traces).
 => HbA2 [ alpha2, delta2 ] (3%) => HbF [ alpha2, gamma2 ] (traces).")
28
Syndromes thalassémiques
Insuffisance de production d’une chaîne de globine = Thalassémie a : défaut de production de chaînes a Thalassémie b : défaut de production de chaînes b La gravité de la maladie est liée au déséquilibre du ratio a/non a, car ce sont les chaînes célibataires qui provoquent Les anomalies membranaires L’accélération de l’apoptose

29
les thalassémies HbA [ alpha2, beta2 ] ↓↓↓ HbF [ alpha2, gamma2 ] ↑↑
Dans la beta thalassémie: HbA [ alpha2, beta2 ] ↓↓↓ HbF [ alpha2, gamma2 ] ↑↑
![les thalassémies HbA [ alpha2, beta2 ] ↓↓↓ HbF [ alpha2, gamma2 ] ↑↑](http://slideplayer.fr/slide/3219251/11/images/29/les+thalass%C3%A9mies+HbA+%5B+alpha2%2C+beta2+%5D+%E2%86%93%E2%86%93%E2%86%93+HbF+%5B+alpha2%2C+gamma2+%5D+%E2%86%91%E2%86%91.jpg "Dans la beta thalassémie: HbA [ alpha2, beta2 ] ↓↓↓ HbF [ alpha2, gamma2 ] ↑↑")
30
Le mécanisme de l’anémie
Principalement: la dysérythropoïèse / destruction des érythroblastes médullaires. secondairement : l’hémolyse périphérique.

31
Les syndromes b-thalassémiques
Selon le degré de l’anémie, et les besoins transfusionnels, on définit les b-thalassémies majeures (anémie de Cooley) intermédiaires = diagnostic clinique !
 intermédiaires. = diagnostic clinique !")
32
maladies héréditaires de transmission autosomique récessive.
La bêtathalassémie est l’hémoglobinopathie la plus répandue dans le Bassin méditerranéen, au Moyen-Orient et en Asie.

33
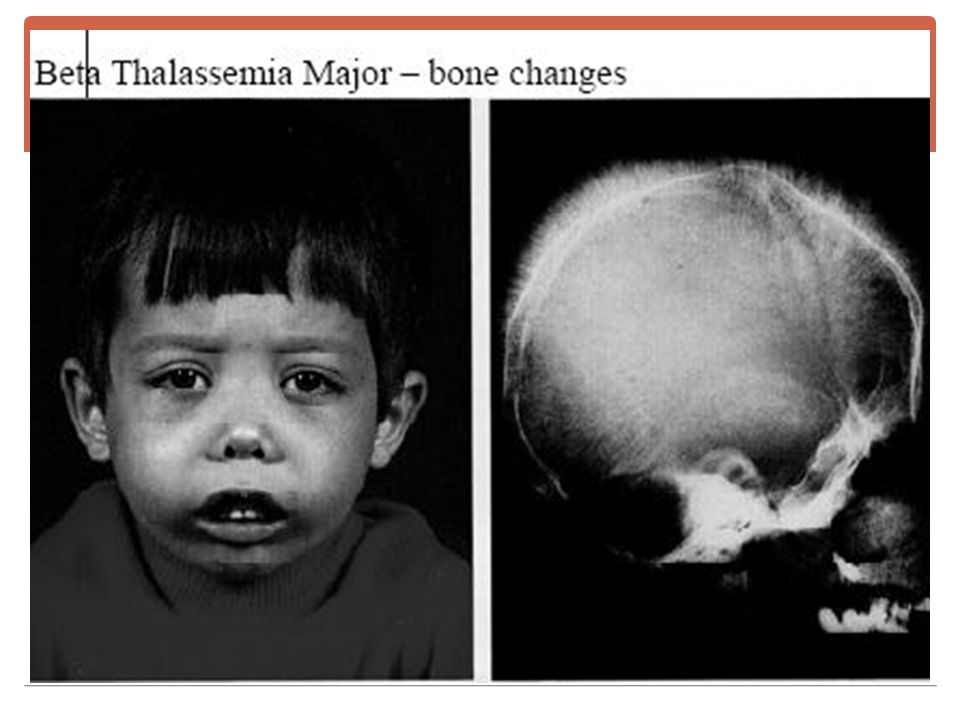
SIGNES CLINIQUES Triade hémolytique Le faciès mongoloïde.
Retard staturo-pondéral.

34
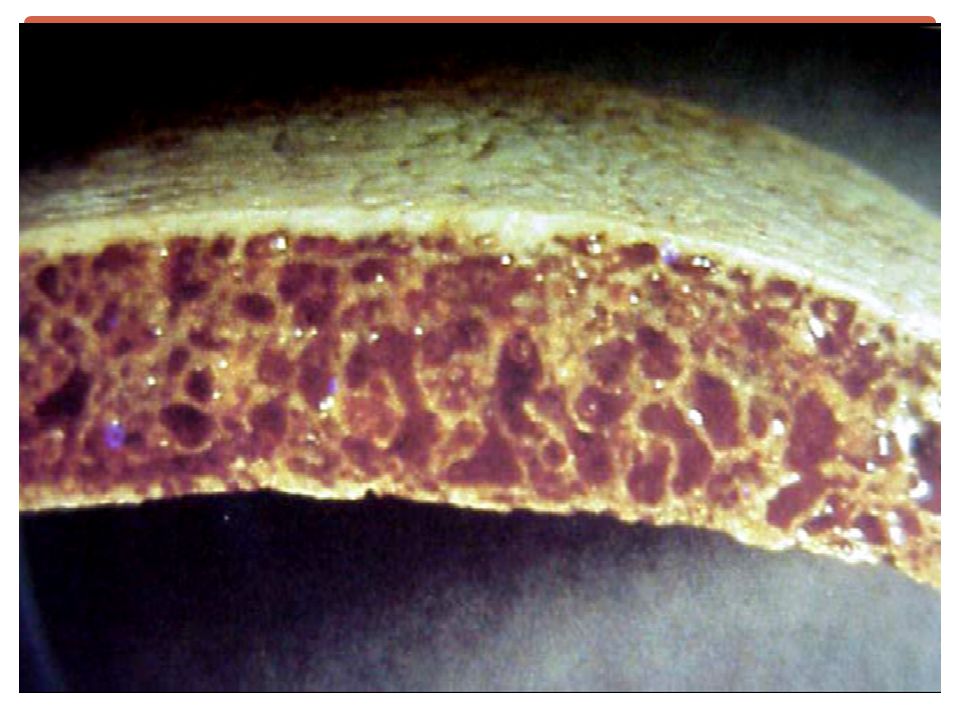
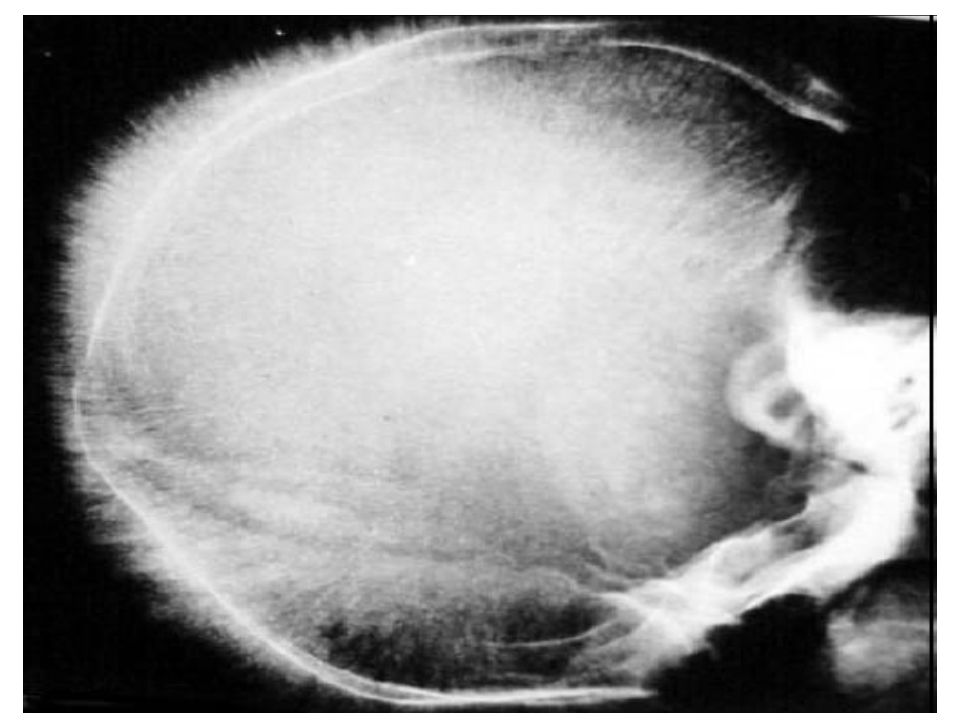
Examens radiologiques
Expansion médullaire / hyperactivité médullaire . Amincissement de la corticale des os longs. Elargissement de la médullaire. Crâne en poil de brosse.

38
Les signes biologiques:
Anémie hypochrome et microcytaire faiblement régénérative. Frottis sanguin = dystrophies importantes des GR. Bilirubine ↑, le fer sérique et la ferritine ↑. l'électrophorèse de l‘Hb : HbF → 100 %, HbA2 = 2 à 7 %

40
Dans les formes majeurs
CORRIGE L'ANEMIE REGIME TRANSFUSIONNEL ASSURER UNE CROISSANCE ET UNE ACTIVITE Nles ET REDUIRE LES MANIFESTATIONS DE LA DYSERYTHROPOEISE RECHERCHER UN DONNEUR HLA IDENTIQUE INTRAFAMILIALE PREVENIR LES COMPLICATIONS DES TF (HEMOSIDEROSE LES DEPISTER PRECOCEMENT TRAITER PRECOCEMENT Le régime transfusionnel systématique associé au traitement chélateur du fer constitue le traitement conventionnel.

41
Thérapie transfusionnelle
L’administration de concentrés de GR déleucocytés,phénotypés RH-KELL, toutes les 3 à 5 semaines, vise à maintenir en permanence le taux d’Hb > 9-10,5 g/dl : (8 à 9 g/dl chez l’adulte).
.")
42
complications des TF Allo-immunisations érythrocytaires → hémolyse retardée. Réactions fébriles immédiates. Réactions allergiques : traitement symptomatique. Complications infectieuses (rares)
")
43
Mécanisme de l’hémosiderose
Dans la TM: principalement / TF ( 1e unité de concentrés globulaires ~ 200 mg de fer.) Dans la TI: principalement / hyperabsorption digestive du fer secondaire à l’anémie.
 Dans la TI: principalement / hyperabsorption digestive du fer secondaire à l’anémie.")
44
Le traitement chélateur du fer
débuté après 10 à 20 transfusions ou lorsque la ferritinémie > μg/l maintenir des concentrations tissulaires en fer n’induisant pas de lésions cellulaire maintenir des ferritinémies ≤ μg/l.

45
3 médicaments ont une AMM dans le TRT de la surcharge transfusionnelle

46
Inconvénients du DFO:

47
Splénectomie Indications: hypersplénisme: besoins TR ≥ 220 cc/Kg/an.
prophylaxie anti-infectieuse: vaccination antipneumococcique au moins 15 jours avant . La couverture vaccinale s’étend au méningocoque, à HIb. L’antigrippale. Antibioprophylaxie prolongée par pénicilline V . toute fièvre élevée → une évaluation médicale en urgence

48
La greffe de cellules souches hématopoïétiques (CSH)
C’est la seule thérapeutique curatrice actuelle. elle est couronnée de succès dans plus de 60 % des cas en Algérie; elle est également moins coûteuse que le traitement à vie conventionnel. La condition : trouver un donneur compatible ou sang du cordon. le taux de réussite de l’intervention dépend de l’âge et de la qualité de la prise en charge.

49
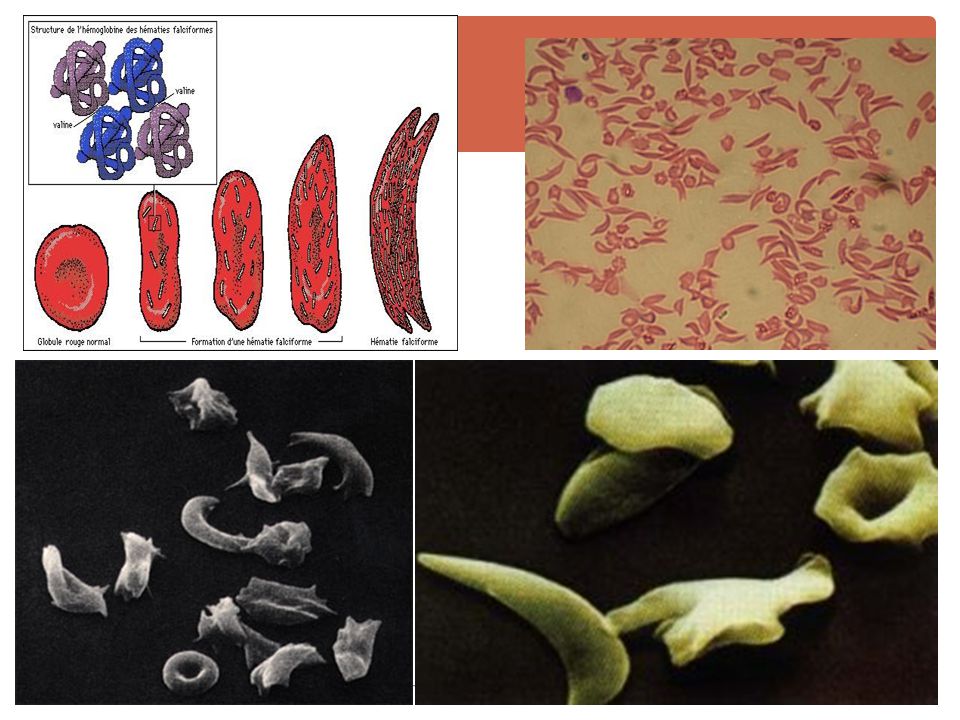
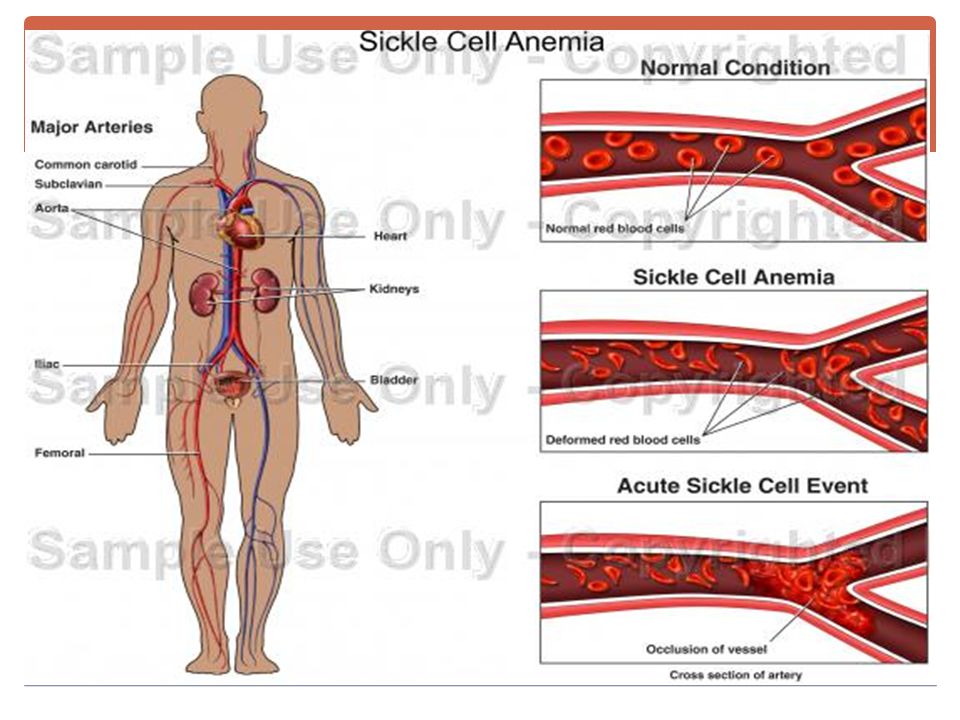
B. La Drépanocytose / hémoglobinose S
Hb S = Hb anle / changement du 6ème AA de la chaîne β : acide glutamique par de la valine. se polymérise / l'hypoxie =» ↓ déformabilité + aspect en faucille = falciformation => microthromboses. Hémolyse intra-vx + extra-vx.

53
Forme homozygote Les manifestations sont variables.
Signes d'anémie hémolytique Crises douloureuses vaso-occlusives Crises de séquestration splénique. Crises d'érythroblastopénie. Lésions viscérales et osseuses : à partir de 15 ans les atteintes dégénératives s’installent (ulcère de jambe, cardiomyopathie, fibrose pulmonaire, rétinite, insuffisance rénale….), Nécrose aseptique de la tête fémorale. L'infection: Les germes : pneumocoque, l'hémophilus influenzae, les salmonelles et le mycoplasme.
, Nécrose aseptique de la tête fémorale. L infection: Les germes : pneumocoque, l hémophilus influenzae, les salmonelles et le mycoplasme.")
54
Les CVO première cause d'hospitalisation.
Variables dans leur type, leur intensité, leur durée, leur fréquence: douleurs osseuses de localisations multiples, une fièvre, signes locaux (douleur à la palpation, œdème). Spontanées ou déclenchées par : une infection, une déshydratation, une acidose, une exposition au froid ou à l'humidité, une hypoxie (voyage en avion, même pressurisé, séjour en altitude, infection respiratoire, anesthésie mal contrôlée). leur répétition = lésions ischémiques des tissus et des organes
. Spontanées ou déclenchées par : une infection, une déshydratation, une acidose, une exposition au froid ou à l humidité, une hypoxie (voyage en avion, même pressurisé, séjour en altitude, infection respiratoire, anesthésie mal contrôlée). leur répétition = lésions ischémiques des tissus et des organes.")
56
Signes biologiques An normocyt normochr régénérative.
GR falciformes sur lame. hyperleucocytose constante. Electrophorèse de l‘Hb : HbS > 50 % . Dc confirmé par le test de falciformation ou test d’emmel sur les GR en milieu pauvre en oxygène.

57
Traitement Vaccination contre le pneumocoque, l'hémophilus et l'hépatite B . Prophylaxie par la pénicilline V: UI / Kg / j. Hyperhydratation. Traiter les CVO : hydratation et analgésiques Transfusion : taux d’Hb doit rester ‹ 10 gr / dl. L'hydréa et la transplantation dans les formes sévéres.

58
Forme hétérozygote Rarement symptomatique. Le diagnostic = test de falciformation + électrophorèse : HbS < 50 %.

59
C. Autres hemoglobinoses :
Hemoglobinose C . L’hemoglobinose D . Les associations : thalassémie et drépanocytose = double hétérozygote = tableau moins sévère.

60
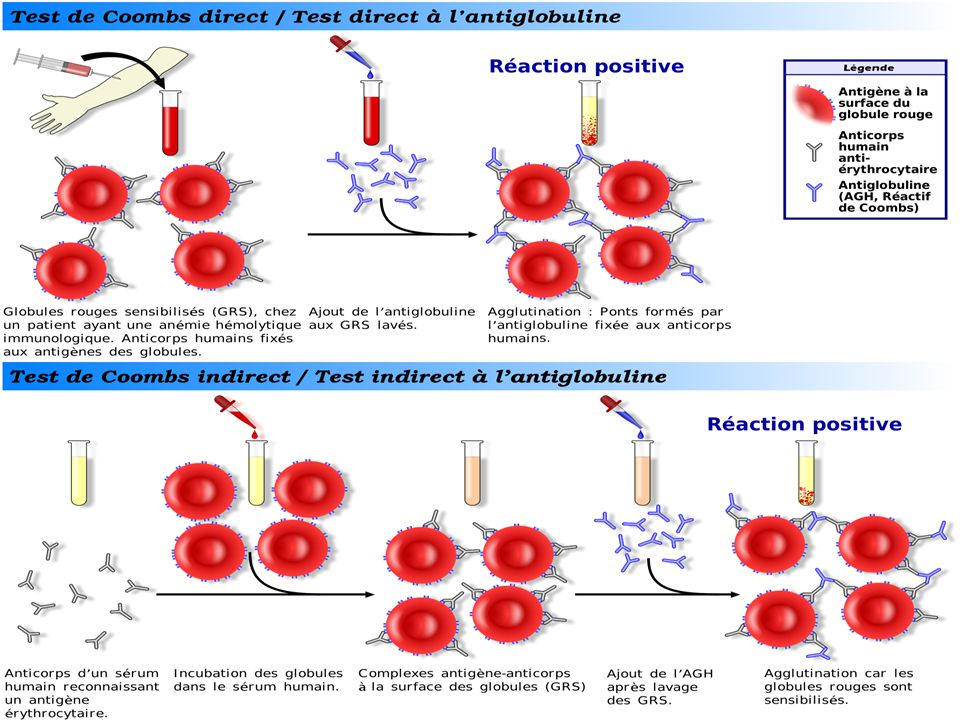
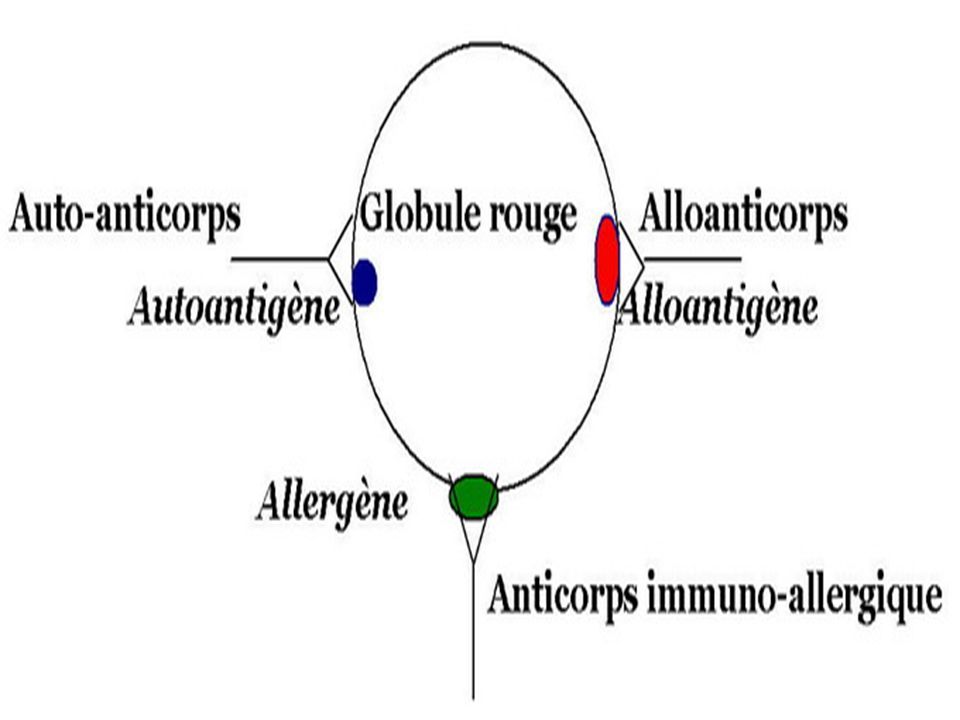
A.H. EXTRA- CORPUSCULAIRES: acquises : Coombs [+] ou immunologiques.
A.H. CORPUSCULAIRES : constitutionnelles A.H. EXTRA- CORPUSCULAIRES: acquises : Coombs [+] ou immunologiques. Coombs [ ̶ ]. A.H. MIXTES.
![A.H. EXTRA- CORPUSCULAIRES: acquises : Coombs [+] ou immunologiques.](http://slideplayer.fr/slide/3219251/11/images/60/A.H.+EXTRA-+CORPUSCULAIRES%3A+acquises+%3A+Coombs+%5B%2B%5D+ou+immunologiques..jpg "A.H. CORPUSCULAIRES : constitutionnelles. A.H. EXTRA- CORPUSCULAIRES: acquises : Coombs [+] ou immunologiques. Coombs [ ̶ ]. A.H. MIXTES.")
62
Anémies hémolytiques immunologiques
1. Accidents transfusionnels 2. Anémie par IMF. 3. Anticorps immunoallergiques 4. Anémies hémolytiques auto-immunes

64
Anémies hémolytiques non immunologiques
Infectieuses Toxiques SHU Anémies hémolytiques des prothèses cardiaques

66
Syndrome hémolytique et urémique
IRA Signes digestifs : diarrhées, nausées, vomissements HTA fréquente +/- Fièvre modérée Dc: Anémie Hémolytique + Schizocytes. Thrombopénie. Insuffisance rénale aiguë. Post diarrhéique de l’enfant : E coli O157:H7

67
Conseil génétique lors des hémoglobinopathies Il faut expliquer aux parents les perspectives de prévention soit par la limitation des naissances. Diagnostic anténatal voir préimplantatoire. Intérêt de l’enquête familiale (Electrophorèse de l’Hb) des parents, fratrie, oncles et cousins…lors des hémoglobinopathies afin de dépister les hétérozygotes et prévenir leur union ultérieure.
 des parents, fratrie, oncles et cousins…lors des hémoglobinopathies afin de dépister les hétérozygotes et prévenir leur union ultérieure.")
Présentations similaires







 Hôpital Saint Louis.>")














 Envahissement médullaire puis systématisé par prolifération de cellules hématopoïétiques malignes ETIOLOGIES 1. Idiopathiques Dans.>")



