Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Les syndromes de prédisposition héréditaire au cancer
Irina Giurgea Hôpital Henri Mondor

2
Les syndromes de prédisposition héréditaire au cancer
Identification de nombreux gènes de prédisposition au cancer 5 à 10% des cancers du sein et du colon sont associés à un syndrome de prédisposition héréditaire au cancer Transmission autosomique dominante d’une mutation germinale d’un gène de susceptibilité Tests génétiques chez les sujets atteints de cancer = cas index Si une mutation est identifiée, tout membre adulte de la famille pourra connaître son statut génétique en réalisant un test prédictif médecine préventive

3
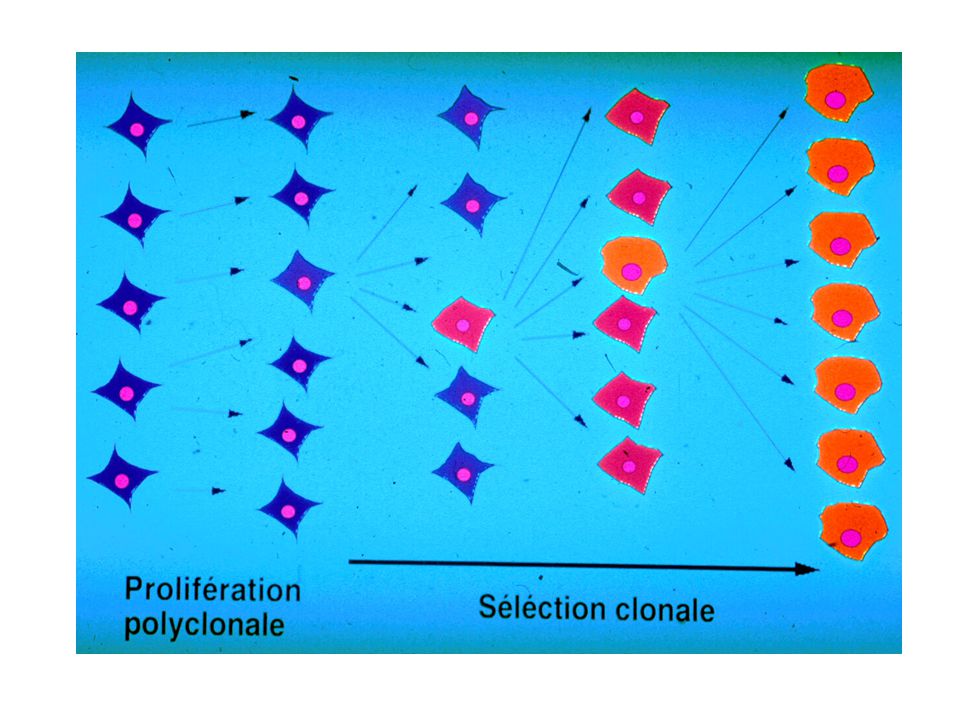
Le mécanisme général Pour un organisme pluricellulaire, le rythme de division doit être contrôlé Ce contrôle de la prolifération est programmé génétiquement La mutation d’un gène contrôlant cette prolifération peut induire un cancer 1016 divisions cellulaires au cours de la vie Taux de mutation spontané pour un gène par division : 10-6 1010 probabilités de mutations par gène!!! Déséquilibre : divisions/mutations/réparations de moins en moins favorable au cours du temps (avec l’âge)
")
4
Les gènes cibles des cancers
Gènes impliqués dans la fidélité (précision de réplication, réparation de l’ADN) le contrôle de la division cellulaire la régulation de la mort cellulaire (apotose) la différenciation cellulaire
 le contrôle de la division cellulaire. la régulation de la mort cellulaire (apotose) la différenciation cellulaire.")
5
incontrôlée (oncogènes)
Croissance incontrôlée (oncogènes) Perte de contrôle prolifération (gènes suppresseurs de tumeurs) Echappement à l’apoptose Activation de l’angiogénèse Invasion tissulaire et métastase Perte de contrôle de la réplication de l’ADN
 Perte de contrôle prolifération. (gènes suppresseurs de tumeurs) Echappement. à l’apoptose. Activation. de l’angiogénèse. Invasion tissulaire. et métastase. Perte de contrôle. de la réplication de l’ADN.")
6
Comment un cancer peut-il être héréditaire?
Localisation des mutations Tissu Somatique Tissu Germinal Pas de transmission à la descendance Transmission à la descendance Cancer spontané non héréditaire Cancer héréditaire

7
Comment savoir que l’on a affaire à un cancer héréditaire ?
établir si transmissibilité selon les lois mendéliennes Enquêtes longues auprès des patients / leur famille On parle de prédispositions familiales à un cancer Cependant, le contexte génique environnemental et extérieur jouera sur l’expression de l’allèle responsable la pénétrance

8
Diversité des présentations de prédisposition aux cancers
Valeur du risque tumoral Nombre d’organes atteints Fréquence de l’atteinte tumorale dans la population Mode de transmission : dominant, récessif, polygénique, interactions gènes - environnement Maladie associée

9
Situations évidentes de prédisposition aux cancers
Risque tumoral élevé Atteinte multifocale Cancer rare dans la population Mode de transmission : dominant Maladie associée

11
Critères permettant retenir des tests de prédisposition génétique aux cancers dans la pratique médicale Risques tumoraux étayés Prise en charge du patient et des apparentés à risque définie Indications des tests définies : essentielle pour les formes familiales de cancer fréquent

12
Objectifs de la consultation de génétique
Répondre à une personne s’interrogeant sur l’origine de son histoire personnelle et familiale de cancers Informer sur les risques tumoraux, en utilisant les ressources de la génétique formelle et de la génétique moléculaire : - > risque élevé ou proche de celui de la population générale Recommander une prise en charge adaptée

13
Première consultation de génétique
1/ Reconstitution de l’histoire personnelle et familiale 2/ Est-on orienté vers un contexte de prédisposition particulier ? 3 / Y-a-t’il d’emblée des recommandations de suivi ? 4/ Y-a-t ’il une indication de test moléculaire ? 5/ Choix du cas index 6/ Information sur les limites et les enjeux des tests

14
Deux types de test génétique
1/ Test du cas index 2/ Test d’un apparenté lorsqu’une mutation a été identifiée

15
Test du cas index 1/ Long, car nécessité de cribler
l’ensemble du ou des gènes impliqués 2/ Signification limitée d’un résultat négatif (pas de mutation identifiée) + Interprétation difficile d’une mutation faux-sens 3/ Étape limitante car si résultat négatif : pas de test possible chez les apparentés
 + Interprétation difficile d’une mutation faux-sens. 3/ Étape limitante car si résultat négatif : pas de test possible chez les apparentés.")
16
Test d’un apparenté 1/ Simple, basée sur la mutation identifiée
chez le cas index 2/ Résultats obtenus en quelques semaines 3/ Signification claire d’un résultat négatif

17
Décret n° du 23 juin 2000 fixant les conditions de prescription et de réalisation des examens des caractéristiques génétiques d’une personne et de son identification - Lois de Bioéthique «Chez une personne asymptomatique mais présentant des antécédents familiaux, la prescription d’un examen des caractéristiques génétiques ne peut avoir lieu que dans le cadre d’une consultation médicale individuelle effectuée par un médecin œuvrant au sein d’une équipe pluridisciplinaire rassemblant des compétences cliniques et génétiques. Cette équipe doit se doter d’un protocole de prise en charge et être déclarée au ministre chargé de la santé. La personne doit être informée des caractéristiques de la maladie recherchée, des moyens de la détecter, des possibilités de prévention et de traitement.»

18
Les syndromes de prédisposition héréditaire au cancer
Les formes familiales de cancer Tumeurs rares : rétinoblastome néphroblastome carcinome médullaire de la thyroïde Tumeurs fréquentes : cancer du sein cancer colo-rectal mélanome malin

19
Rétinoblastome tumeur rare de la rétine
Rétinoblastome D, 12 mois Rétinoblastome G, 14 mois ? ? Rétinoblastome, 2 ans Rétinoblastome, 3 mois tumeur rare de la rétine atteignant un enfant sur

20
Rétinoblastome Dans 90% des cas : pas d’histoire familiale
60% cas unilatéraux, 30% cas bilatéraux Dans 10% des cas : histoire familiale transmission dominante (un des deux parents atteint dans l’enfance) le plus souvent atteinte bilatérale maladie précoce : diagnostic avant 12 mois
 le plus souvent atteinte bilatérale. maladie précoce : diagnostic avant 12 mois.")
22
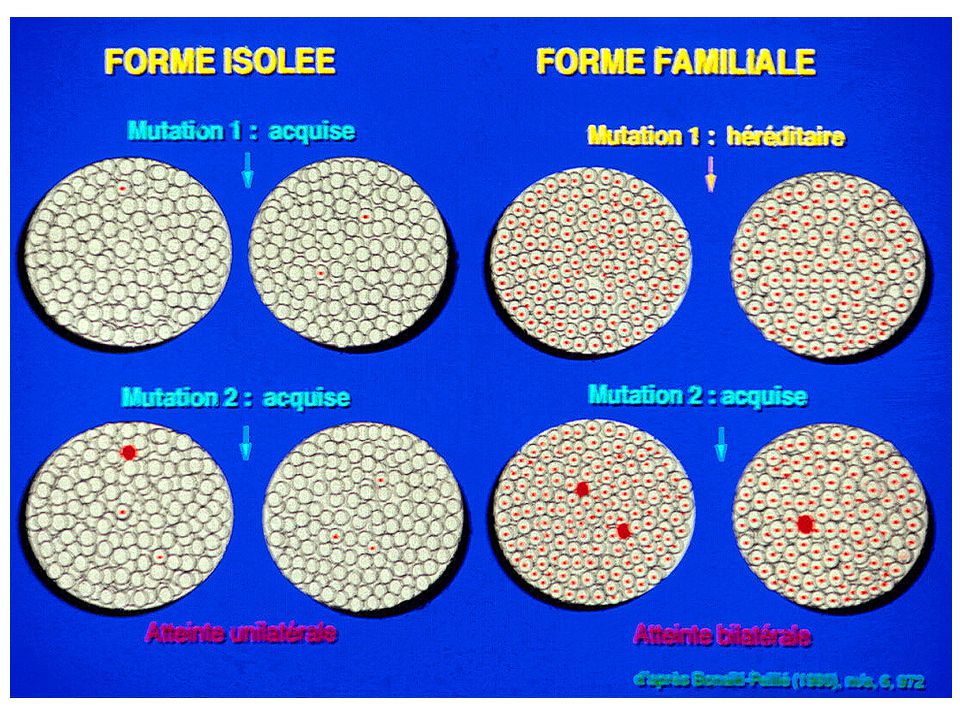
Génétique du Rétinoblastome Modèle de Knudson, 1971
Deux altérations génétiques dans une cellule de la rétine sont nécessaires (mais peut-être pas suffisantes) à la transformation tumorale
 à la transformation tumorale.")
24
Le modèle de Knudson, complété par Comings Identification du gène RB1
1ère mutation somatique 2ème mutation somatique Cas isolé - Tumeurs unilatérales - Survenue tardive Lignée germinale: Pas de mutation Clone cellulaire 1 allèle muté Tumeur 2 allèles mutés - Haute fréquence de tumeurs multiples et bilatérales - Survenue précoce 2ème mutation somatique Cas familial Lignée germinale: 1 allèle muté Tumeur 2 allèles mutés

25
Le gène RB1 27 exons 183 kb Épissage alternatif de l’exon 3
Messager de 4.7 kb avec un cadre de lecture de 2.7 kb Conservé entre les espèces (Rat, souris, coq, fugu…) A B NLS
 A. B. NLS.")
26
Protéine pRB Protéine nucléaire de 110 kDa Expression ubiquitaire
Pocket proteins (p107, p130) Inhibition de facteurs de transcription famille E2F Classon, 2002 pRB inhibe l’expression génique par remodelage chromatinien au niveau des régions promotrices des gènes
 Inhibition de facteurs de transcription famille E2F. Classon, pRB inhibe l’expression génique par remodelage chromatinien au niveau des régions promotrices des gènes.")
27
pRB : fonctions nombreuses
et complexes > 100 protéines d’interaction Classon, 2002

28
Gène RB1 : Pathologie moléculaire
27 1 2 3 4 7 8 9 10 13 17 18 12 16 19 20 21 22 23 25 g.2187insC IVS01-2A>G IVS02+3delA g.42006insAA Q176X IVS05-1G>A IVS06+1G>A (2) R255X R251X IVS08-10T>C E313D E313G IVS09-1G>C R320X(6) Q344X g.64414delA g.70270insA g.70245delT IVS12+1G>T R455X (2) g.76453delG R445X (2) R467X (3) IVS15+17del21 IVS15-2A>G IVS17+5G>A g del4 IVS19+1delG IVS19-2A>G g del15 IVS20+2del4 IVS21+1G>T (2) g insG E746X IVS22-1delG R787X (3) g delC g insT S834X IVS24+1G>A R556X (2) P20L IVS14+1G>A E545X E466X g.2168insG g delG W75X Q563X IVS06+1G>T (2) g.61756delT R331X g.64368delT g delA Del P-17 (2) Del P- 6 Del P-27 (5) Del 2 Del 3 (2) Del 8 Del Del Del P-27 Del P-2 (2) Del Houdayer et al, 2004
 R255X. R251X. IVS08-10T>C. E313D. E313G. IVS09-1G>C. R320X(6) Q344X. g.64414delA. g.70270insA. g.70245delT. IVS12+1G>T. R455X (2) g.76453delG. R445X (2) R467X (3) IVS15+17del21. IVS15-2A>G. IVS17+5G>A. g del4. IVS19+1delG. IVS19-2A>G. g del15. IVS20+2del4. IVS21+1G>T (2) g insG. E746X. IVS22-1delG. R787X (3) g delC. g insT. S834X. IVS24+1G>A. R556X (2) P20L. IVS14+1G>A. E545X. E466X. g.2168insG. g delG. W75X. Q563X. IVS06+1G>T (2) g.61756delT. R331X. g.64368delT. g delA. Del P-17 (2) Del P- 6. Del P-27 (5) Del 2. Del 3 (2) Del 8. Del Del Del P-27. Del P-2 (2) Del Houdayer et al,")
29
RB1 : Présentations individuelles et familiales
RB1 : Présentations individuelles et familiales taux de mutations identifiées Rb familiaux > 95% Rb bilatéraux sporadiques ~ 80% Rb unilateraux 15%

30
Rétinoblastome D, 12 mois Rétinoblastome G, 14 mois ? Rétinoblastome, 3 mois Un risque sur deux de prédisposition Si prédisposé, 90% de risque d’être atteint

31
Prise en charge des enfants à risque de rétinoblastome
Fond d’œil (milieu spécialisé, sous anesthésie générale dans la petite enfance) dès la première semaine de vie tous les mois -> 18 mois tous les 3 mois -> 4 ans tous les 6 mois -> 20 ans Test génétique : recherche de la mutation RB1 responsable chez le cas index puis chez l’enfant indemne Si non porteur de la mutation identifiée dans la famille : levée de la surveillance
 dès la première semaine de vie. tous les mois -> 18 mois. tous les 3 mois -> 4 ans. tous les 6 mois -> 20 ans. Test génétique : recherche de la mutation RB1. responsable chez le cas index. puis chez l’enfant indemne. Si non porteur de la mutation identifiée dans la famille : levée de la surveillance.")
32
Délétion de 1 nucléotide en position 380
Mutation du gène RB1 Délétion de 1 nucléotide en position 380 ?

33
Cancer du sein

34
? ? ? ? ? T foie, 80 T poumon, 70 T sein, 65 T col utérus, 40
T ORL, 70 T sein, 75 ? ? ? ? T sein, 55 T côlon, 65 65 ans

35
T sein, 45 T sein, 56 T sein, 54 T sein, 42 ? T sein, 36 T sein, 34 âgée 39 ans T sein, 40

36
BRCA1 BRCA2 17q21 2cM 13q12 King, 1990 BCLC, 1993 Skolnick, 1994
Stratton, 1994 Stratton, 1995

37
Réparation Ubiquitinylation BRCA1 : Fonctions Accès à l ’ADN
Remodelage de la chromatine HDACs SW1/SNF H2AX Ub BRG1 Réparation P ATM-ATR CHK2 kinase Dommage de l ’ADN Recombinaison Homologue RAD51 BRCA2 RB PLK1 Arrêt en phase S et G2 Régulation checkpoint CHK1 P53 WAF1 GADD45 Phase G1/S Phase G2/M Ub FANCD2 BARD1 Cibles Ubiquitinylation ? BAP1 Non homologous end Joining MRE11-RAD50-NBS1 MSH2-MSH6 BLM BRCA1 Ub Réparation couplée à la transcription RNA polII Ub Régulation de la transcription

38
Phénotype cellulaire des hétérozygotes BRCA1+/-
Hypersensiblité aux radiations ionisantes - induction de micronoyaux - survie cellulaire Défaut de fidélité de réparation des CDB de l’ADN - test de Host-Cell End-Joining

39
Variabilité inter et intra familiale du risque tumoral
BRCA1 : 185delAG A : Famille sein-seul B : Famille ovaire-seul C : Famille sein-ovaire dg 69 sein 46 ans Dg 55 ovaire dg 60 sein dg 82 sein dg 50 dg 45 ovaire dg 41 sein dg 60 sein 73 ans dg 41 ovaire dg 48 dg 49 sein 65 ans dg 40 ovaire dg 47 ovaire dg 39 sein

40
A quelles situations familiales correspond une probabilité de prédisposition > ~ 25 % ?
1/ Au moins trois cas T sein ou ovaire, unis entre eux par un lien de premier ou second degré, appartenant à la même branche parentale, quelque soit l’âge au Dg 2/ Deux cas T sein unis entre eux par un lien de premier degré dont - 1 cas avant 40 ans ou - 1 cas avant 50 ans et 1 cas avant 70 ans ou - 1 cas masculin 3/ Un cas T ovaire, quelque soit l’âge au dg , et un cas T sein chez un premier degré

41
Indications consultation de génétique = étude génétique cas index
Introduction d’un score T sein chez une femme avant 30 ans 4 T sein chez une femme entre 30 et 39 ans 3 T sein chez une femme entre 40 et 49 ans 2 T sein chez une femme entre 50 et 70 ans 1 T sein chez un homme T ovaire Addition de chaque point par cas T sein (ou ovaire) si cas dans la même branche parentale. 5 : excellente indication 3 ou 4 : indication possible 1 ou 2 : utilité médicale faible
 si cas dans la même branche parentale. 5 : excellente indication. 3 ou 4 : indication possible. 1 ou 2 : utilité médicale faible.")
42
T sein, 56 T sein, 45 T sein, 56 T sein, 42 âgée 39 ans T sein, 36 T sein, 34 T sein, 40 Cas index

43
prostate, colon,mélanome pancréas
Risques de cancers associés à BRCA cancer du sein 80% cancer du sein 5% cancer de l’ovaire 20-40% autres cancers prostate, colon,mélanome pancréas D’après ASCO

44
Surveillance des femmes BRCA
Mesures Fréquence Age de début Examen seins 2-3 fois par an 25 ans Mammographie 1 fois par an 30 ans (ou 5-10 ans av dg le plus précoce) Echographie 1-2 fois par an 25-30 ans IRM (protocole) 1 fois/an 25 ans 2 fois /an 35 ans Examen clinique ovaires Toutes ces mesures sont basées sur le fait que la détection précoce d’une lésion tumorale permet un traitement optimal à un stade précoce en vue d’une meilleure chance de guérison. Ce qui n’est pas prouvé pour les BRCA. En fait ces recommandations sont extrapolées de celles de la population générale. Si bien que des différences existent entre les américains et les européens notamment en ce qui concerne l’autopalpation des seins, non recommandée en France en raison de l’anxiété qu’elle peut générer, et la chirurgie prophylactique. Ce dernier point pouvant être seulement le reflet des différences socio-culturelles. Dans le futur, place du lavage ductal, Doppler contraste, mammographie contraste, electropotentiels etc… Quelque soient les méthodes utilisées, le problème reste celui des cancers de l’intervalle surtout avant 50 ans. Tumeurs moins visibles à la mammographie chez les BRCA? En fait, les tumeurs indifférenciées de grade III seraient moins visibles même chez les non-BRCA L’échographie n’a pas du tout été évalué parmi les femmes BRCA 1-2 fois/an 35 ans Echo endovag Doppler couleur CA 125 1-2 fois/an 40 ans ?
 Echographie. 1-2 fois par an ans. IRM (protocole) 1 fois/an. 25 ans. 2 fois /an. 35 ans. Examen clinique. ovaires. Toutes ces mesures sont basées sur le fait que la détection précoce d’une lésion tumorale permet un traitement optimal à un stade précoce en vue d’une meilleure chance de guérison. Ce qui n’est pas prouvé pour les BRCA. En fait ces recommandations sont extrapolées de celles de la population générale. Si bien que des différences existent entre les américains et les européens notamment en ce qui concerne l’autopalpation des seins, non recommandée en France en raison de l’anxiété qu’elle peut générer, et la chirurgie prophylactique. Ce dernier point pouvant être seulement le reflet des différences socio-culturelles. Dans le futur, place du lavage ductal, Doppler contraste, mammographie contraste, electropotentiels etc… Quelque soient les méthodes utilisées, le problème reste celui des cancers de l’intervalle surtout avant 50 ans. Tumeurs moins visibles à la mammographie chez les BRCA En fait, les tumeurs indifférenciées de grade III seraient moins visibles même chez les non-BRCA. L’échographie n’a pas du tout été évalué parmi les femmes BRCA. 1-2 fois/an. 35 ans. Echo endovag. Doppler couleur. CA fois/an. 40 ans")
45
Mastectomie prophylactique
POUR efficacité prouvée cancers de l’intervalle de la surveillance standard absence de lésions pré-cancéreuses chimioprévention non démontrée mauvais pronostic (?) des cancers du sein BRCA esthétique meilleure CONTRE geste « coûteux » et irréversible amélioration du pronostic des cancers du sein chimiosensibilité des cancers BRCA interventions multiples
 des cancers du sein BRCA. esthétique meilleure. CONTRE. geste « coûteux » et irréversible. amélioration du pronostic des cancers du sein. chimiosensibilité des cancers BRCA. interventions multiples.")
46
Ovariectomie prophylactique
POUR Surveillance peu efficace Efficacité prouvée Geste peu morbide Quand? CONTRE Risque résiduel Ménopause précoce À partir de 35 ans si projet parental accompli, ou à partir de 40 ans (BRCA1), 50 ans (BRCA2)
, 50 ans (BRCA2)")
47
Cancer du colon

48
Syndrome H.N.P.C.C. HISTORIQUE
1913- AS Warthin: Description d ’une forme « héréditaire » de cancer colo-rectal dans une grande famille (famille G.) 1966- HT Lynch: « cancer family syndrome » à propos de deux grandes familles 1971- HT Lynch: Famille G 1984- CR Boland: introduction du sigle HNPCC pour: « hereditary non polyposis colo-rectal cancer » 1991- Critères d ’Amsterdam
 HT Lynch: « cancer family syndrome » à propos de deux grandes familles HT Lynch: Famille G CR Boland: introduction du sigle HNPCC pour: « hereditary non polyposis colo-rectal cancer » Critères d ’Amsterdam.")
49
HNPCC

50
Le cancer héréditaire du colon HNPCC (Hereditary Non Polyposis Colorectal Cancer)
1/200 dans le monde occidental Hérédité - Autosomique dominante – risque de ½ de transmettre / recevoir la mutation Les porteurs ont un risque important de développer le cancer avant 50 ans 6 gènes sont impliqués dans la réparation des mésappariements de l’ADN (MMR=mismatch repair): MLH1 (30 %), MSH2 (40%), MSH6 (< 5%), MLH3, PMS1/2
: MLH1 (30 %), MSH2 (40%), MSH6 (< 5%), MLH3, PMS1/2.")
51
Le cancer héréditaire du colon HNPCC (Hereditary Non Polyposis Colorectal Cancer)
Anomalies des séquences d’ADN répétitives dites microsatellites par PCR Immunohistochimie perte d’expression de la protéine muté hMLH1 hMSH2

52
Fonction du gène MSH2 (HNPCC)
MSH2: MutS Human homologue n°2 mutS d’ Escherichia coli et hexA de Streptococcus pneumoniae L’inactivation de ces gènes confèrent une augmentation de la fréquence de mutations allant jusqu’à un facteur 100. Famille de gènes intervenant dans la correction des erreurs de réplication que l’on retrouve dans toutes les espèces et tous les règnes du vivant. L’équilibre du maintien du patrimoine génétique est rompu!

53
Pourquoi la perte de fonction est-elle dominante d’un point de vue cancer?
Une cellule hétérozygote pour MSH2 répare les erreurs. De ce point de vue, la perte de fonction de MSH2 est récessive. MSH2 Homozygote [sain] Heterozygote [cellule saine] Mutation 1 Msh2 Ce n’est pas la cellule hétérozygote qui déclenche le cancer mais l’homozygote mutant! 70% par recombinaison mitotique 30% par mutation aléatoire [cellule hypermutatrice] Mutation 2 msh2’ msh2

54
Le syndrome HNPCC Les tumeurs HNPCC sont dans 95 % des cas RER + .
1/3 des K du côlon droit sont RER + vs 5% pour le côlon gauche. 15 à 20 % des K sporadiques ont une perte d’expression de MLH1 par méthylation du promoteur lié à l’âge (Kakar et al, Cancer 2003) 0 % ≤ 40 ans 4,9 % entre 41 et 50 ans 8,3 entre 51 et 60 ans 13% > 60 ans
 0 % ≤ 40 ans. 4,9 % entre 41 et 50 ans. 8,3 entre 51 et 60 ans. 13% > 60 ans.")
55
Syndrome HNPCC Critères d ’Amsterdam II 1999
Agrégation familiale d ’au moins 3 cas de cancers appartenant au spectre étroit prouvés histologiquement liés deux à deux au 1er degré > polypose colo-rectale exclue > touchant 2 générations distinctes Diagnostiqué avant 50 ans dans au moins un cas. Critères d’Amsterdam II élargis Au minimum 2 sujets atteints de cancers appartenant au spectre étroit apparentés au 1er degré

56
Le syndrome HNPCC Les risques tumoraux cumulés en % à 70 ans
HOMMES FEMMES CCR 50 à 60% 30 à 40% ENDOMETRE à 40% ESTOMAC 10% 10 % GRELE (1 étude à 4%) 1 à 4 % 1 à 4% VOIES BILIAIRES 5 % 5% VOIES URINAIRES 5% 5% OVAIRES % Pas de surrisque pour K poumon, prostate et sein Syndrome de TURCOT: + GB Syndrome de Muir-Torre: + CARCINOME SEBACES
 1 à 4 % 1 à 4% VOIES BILIAIRES 5 % 5% VOIES URINAIRES 5% 5% OVAIRES 8 % Pas de surrisque pour K poumon, prostate et sein. Syndrome de TURCOT: + GB. Syndrome de Muir-Torre: + CARCINOME SEBACES.")
57
Test d’un cas atteint A tester pour rechercher une mutation familiale inconnue = cas index Côlon dx 42 Côlon dx 38 d.45 Côlon dx 45 Proposant = 1ère personne vue en consultation

58
Surveillance Clinique pour les « Porteurs » De Mutation Germinale (HNPCC)
Site Procédure Age début Intervalle Côlon Coloscopie (>18) 2 Endomètre Examen gynécologique, (et ovaires) échographie transvaginale hystéroscopie et biopsies CA 125 Estomac Gastroscopie Appareil Echographie Urinaire Cytologie urinaire UIV
 2. Endomètre Examen gynécologique, (et ovaires) échographie transvaginale hystéroscopie et biopsies. CA 125. Estomac Gastroscopie Appareil Echographie Urinaire Cytologie urinaire. UIV 2.")
59
Recommandations pour les « Porteurs » de Mutation Germinale (HNPCC)
Exérèse systématique des polypes Hystérectomie, ovariectomie Si cancer colique ou adénome >1cm : colectomie totale avec anastomose iléo-rectale surveillance par rectoscopie annuelle Chimioprévention : aspirine/amidon (essai en cours)
")
60
Objectifs de la consultation de génétique
Répondre à une personne s’interrogeant sur l’origine de son histoire personnelle et familiale de cancers Informer sur les risques tumoraux, en utilisant les ressources de la génétique formelle et de la génétique moléculaire : - > risque élevé ou proche de celui de la population générale Recommander une prise en charge adaptée

61
Première consultation de génétique
1/ Reconstitution de l’histoire personnelle et familiale 2/ Est-on orienté vers un contexte de prédisposition particulier ? 3 / Y-a-t’il d’emblée des recommandations de suivi ? 4/ Y-a-t ’il une indication de test moléculaire ? 5/ Choix du cas index 6/ Information sur les limites et les enjeux des tests

62
Deux types de test génétique
1/ Test du cas index 2/ Test d’un apparenté lorsqu’une mutation a été identifiée

63
Test du cas index 1/ Long, car nécessité de cribler
l’ensemble du ou des gènes impliqués 2/ Signification limitée d’un résultat négatif (pas de mutation identifiée) + Interprétation difficile d’une mutation faux-sens 3/ Étape limitante car si résultat négatif : pas de test possible chez les apparentés
 + Interprétation difficile d’une mutation faux-sens. 3/ Étape limitante car si résultat négatif : pas de test possible chez les apparentés.")
64
Résultats positifs cas index
Implication pour le traitement Evaluation risque autres cancers Risque de transmission descendance Information aux apparentés

65
Résultats négatifs cas index : pourquoi?
Ce n’est pas une prédisposition héréditaire Limites techniques des tests (manque de sensibilité, mutations introniques ou du promoteur) Réarrangements complexes: techniques en cours d’évaluation Variants rares et inconnus: tests fonctionnels à développer Autres gènes Conséquences : pas de test familial disponible
 Réarrangements complexes: techniques en cours d’évaluation. Variants rares et inconnus: tests fonctionnels à développer. Autres gènes. Conséquences : pas de test familial disponible.")
66
Test d’un apparenté 1/ Simple, basée sur la mutation identifiée
chez le cas index 2/ Résultats obtenus en quelques semaines 3/ Signification claire d’un résultat négatif

67
Décret n° du 23 juin 2000 fixant les conditions de prescription et de réalisation des examens des caractéristiques génétiques d’une personne et de son identification - Lois de Bioéthique «Chez une personne asymptomatique mais présentant des antécédents familiaux, la prescription d’un examen des caractéristiques génétiques ne peut avoir lieu que dans le cadre d’une consultation médicale individuelle effectuée par un médecin œuvrant au sein d’une équipe pluridisciplinaire rassemblant des compétences cliniques et génétiques. Cette équipe doit se doter d’un protocole de prise en charge et être déclarée au ministre chargé de la santé. La personne doit être informée des caractéristiques de la maladie recherchée, des moyens de la détecter, des possibilités de prévention et de traitement.»

68
Résultats positifs cas index
Implication pour le traitement Evaluation risque autres cancers Risque de transmission descendance Information aux apparentés

69
Test pré-symptomatique Mutation familiale identifiée
Après 18 ans Consultation: information sur les risques de cancer, règles de transmission à la descendance, moyens de prévention, dépistage… Deux prélèvements sanguins à distance Suivi psychologique

70
Résultats test pré-symptomatique
Risques de la population générale Suivi médical fonction de l’histoire personnelle Evaluation risques individuels Dépistage, prévention Suivi psychologique Etudes de cohorte, facteurs modificateurs Résultat négatif Résultat positif

71
Des tests génétiques mais pas n’importe comment !
Conclusion Des tests génétiques mais pas n’importe comment ! Équipe pluridisciplinaire Protocole de prise en charge Information complète, compréhensible Autonomie de décision du candidat au test -> Consentement libre et éclairé

Présentations similaires







>")
 Pr E. Tournier-Lasserve>")


 Pr E. Tournier-Lasserve>")








>")