Télécharger la présentation
1
PHARMACOCINETIQUE Pr.Houda Filali
Laboratoire de Biochimie-Pharmacologie CHU Ibn Rochd Service de Pharmacologie-Toxicologie FMPC

3
Pharmacocinétique Etude du devenir des médicaments dans l'organisme.
Etude de la chronologie de l'effet.

4
Différentes voies d'administration d'un médicament
voie orale ou per os voie intra-veineuse voie sub-linguale voie rectale voie sous-cutanée voie cutanée ou trans-dermique voie intra-musculaire dans un organe ou in situ : intra-oculaire, intra-thécale, intra-tumoral… voie nasale ou oculaire (collyres) voie inhalée
 voie inhalée.")
5
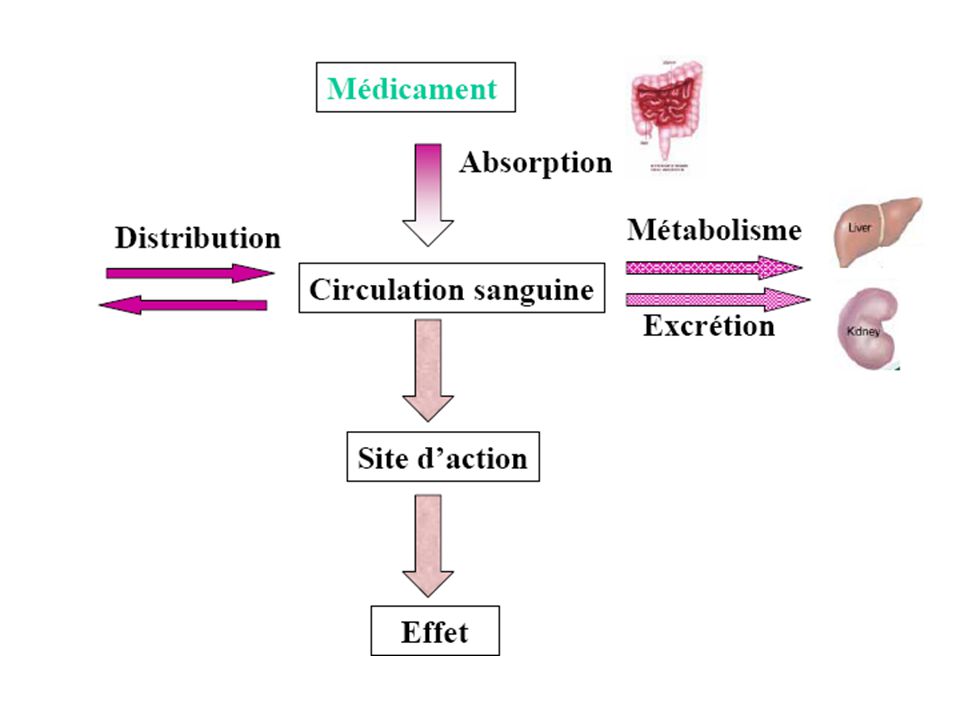
Absorption et Distribution Elimination Administration
S.Porte Foie Urines Peros Rectal Reins Intestins Selles Percut Peau PLASMA IV Sein Lait IM Muscle Intrathécal Cerveau Placenta Foetus Inhalation Poumon Air expiré

6
Pharmacocinétique - Formes galéniques - Voies d’administrations
- Posologies - Rythme d’administration

7
ADME Absorption Distribution Métabolisme Élimination

8
Absorption 1- Etape biopharmaceutique ( voies extra vasculaires)
2- Etape de résorption : Passage transmenbranaire

9
FORME PHARMACEUTIQUE SOLIDE PRINCIPE ACTIF EN SOLUTION
Absorption 1- Etape biopharmaceutique FORME PHARMACEUTIQUE SOLIDE désintégration AGREGATS désagrégation PARTICULES dissolution PRINCIPE ACTIF EN SOLUTION RESORPTIION

10
Absorption 2- Étape de résorption : Passage transmembranaire ++++
- Diffusion passive Diffusion facilitée Diffusion active Autres….

11
Caractéristique du médicament : Caractéristique du patient
Absorption Caractéristique du médicament : Voie d’administration Forme galénique Ionisation Liposolubilité Caractéristique du patient pH site d’absorption Temps de transit Surface absorbante Circulation sanguine Âge Pathologie

12
Absorption Paramètre quantificatif de l’absorption ?

13
La biodisponibilité fraction de la dose administrée qui atteint la circulation générale et vitesse d’accession à la circulation générale

14
Biodisponibilité AUC = Aire sous la courbe Concentration
AUC =ASC = S [ C ] x dt Biodisponibilité = AUC peros AUC IV AUC = Aire sous la courbe Biodisponibilité Concentration Temps IV Peros

15
La biodisponibilité Absolue
- comparaison des autres voies d’administration à la voie veineuse : choix forme d’administration Relative comparaison de deux formulations: bioéquivalence pour la mise sur le marché de génériques comparaison de deux voies d’administration (Autres que IV)
")
16
La biodisponibilité Biodisponibilités absolue et relative

17
Pourquoi connaître la biodisponiblité absolue ?
Pour déterminer l’exposition de l’organisme au médicament Dose par unité de temps = Clairance x Conc.efficace Biodisponibilité

18
Inconvénient d’une faible biodisponibilité absolue
Sur-exposition (effets indésirables) AUC ou concentrations Seuil toxique Exposition Seuil thérapeutique Sous-exposition (échec thérapeutique, résistance) 1 5 Dose
 AUC ou. concentrations. Seuil. toxique. Exposition. Seuil. thérapeutique. Sous-exposition. (échec thérapeutique, résistance) Dose.")
19
La biodisponibilité Importance de la vitesse d’absorption CORTICOÏDES
ANTIBIOTIQUES Fluoroquinolones Aminoglycosides Beta-lactamines

20
Biodisponibilité - quantification
Excellente entre 80 et 100% Bonne entre 60 et 80% Moyenne entre 40 et 60% Mauvaise si < 40%

21
Effet de premier passage
ABSORPTION Veine porte Paroi intestinale foie Lumière intestinale Circulation générale intestinal hépatique fèces METABOLISME

23
Elimination intestinale
Effet de 1er passage Elimination dans la paroi intestinale : « Premier passage » (métabolisme), excrétion (P-gp) Veine porte FOIE Circulation générale Intestin Elimination hépatique « Premier passage » Elimination intestinale
, excrétion (P-gp) Veine porte. FOIE. Circulation générale. Intestin. Elimination hépatique « Premier passage » Elimination intestinale.")
24
Absorption digestive ENTEROCYTE Veine porte Résorption CYP450 P-gp
Principe actif Principe actif CYP450 P-gp Métabolites inactifs ENTEROCYTE

25
Cycle entérohépatique
Foie Veine porte Estomac Voies biliaires

26
3. La voie rectale (2)
")
27
Pharmacocinétique Absorption Distribution Métabolisme Élimination

28
Distribution Fixation aux protéines plasmatiques Par des liaisons
Hydrogène Ioniques Électrostatiques faibles Hydrophobes Réversible En équilibre SEULES LES FORMES LIBRES SONT ACTIVES P:46

29
Protéines sériques 48 % Erythrocytes 51 % Intérieur 35 % Membranes
16 % Libre 1.4 % Erythrocytes 51 %

30
Liaison aux protéines Médicament libre + Protéine libre <----> Complexe M + P La forme ionisée Albumine surtout Interaction par compétition En pratique, attention si fixation élevée et index thérapeutique faible

31
Fixation aux protéines
Fortement fixés si fixation > 75% Moyennement fixés si fixation comprise entre 40 et 75% Faiblement fixés si fixation < 40%

32
La distribution Affinité tissulaire: Liposolubilité - pH
Vascularisation Volume Redistribution Compartiment Volume de distribution

33
Volume apparent de distribution
Facteurs déterminant la diffusion tissulaire C M Vd = QM Ci Ci t

34
Volume apparent de distribution
Plasma : facilement mesurable Tissulaire / Interstitiel Cellulaire

35
Volume apparent de distribution
Dose X Biodisponibilité Vd = Concentration sérique

36
Volume apparent de distribution
Concentration tissulaire : dans le plasma Vd : élevé Hydrosoluble : dans le plasma Vd : faible Liposoluble :

37
Volume de distribution - Quantification
Petit si < 1 L/kg Moyen si = 1L/kg Grand si > 1L/kg

38
Ce qui modifie la distribution
Age Obésité = graisses Hémodynamique Hydratation Albuminémie

40
Modèle bi-compartimental
Plasma Tissus vascularisés Autres tissus

41
Distribution du médicament
La barrière capillaire PM < 64000 F libre La barrière hémoméningée = barrière lipidique Liposoluble non ionisé au pH plasmatique La barrière placentaire PM entre 500 et 1000

42
Volumes liquidiens de l’organisme
Homme de 7O kg eau totale: 60% > 42l volume intracellulaire: 40% > 28l volume extracellulaire: 20% > 14l volume plasmatique: 5% > 3,5l

43
Question: VD? Médicament hydrosoluble et PM élevé ? VD = …….
Médicament Lipophile et PM petit ? VD = ……. VD = index de fixation tissulaire du médicament

44
Application en Réanimation
Epuration extrarénale: - médicament peu lié aux protéines - volume de distribution petit

45
Ce qu’il faut retenir Seule la fraction libre est active
La durée d’action d’un médicament est proportionnelle à son degré de liaison aux protéines sanguines

46
Ce qu’il faut retenir Pour passer une barrière cellulaire, le médicament doit être: LIPOSOLUBLE NON IONISE LIBRE, non lié aux protéines.

47
Pharmacocinétique Absorption Distribution Métabolisme Élimination

48
Métabolisme Biotransformation : Ensemble des réactions biochimiques que subissent les substances endogènes et exogènes résultant en une diminution de leur caractère lipophile lui permettant une excrétion facilitée

49
Métabolisme

50
Métabolisme Site FOIE : Principal organe de biotransformation + Débit sanguin important INTESTIN : Activité du CYP3A4 démontrée POUMONS : Induction du CYP1A1 par la fumée de cigarette PEAU, REIN, CERVEAU… : Faibles concentrations en P450 Objet PK ==> Produits hydrosolubles >>> urines + bile PD ==> Le métabolisme peut être qualitativement important par la nature des métabolites produits, toxiques, actifs

51
Métabolisme veine hépatique artère hépatique (sang oxygéné)
veine porte hépatique (sang désoxygéné) artère hépatique (sang oxygéné)
 artère hépatique. (sang oxygéné)")
54
Métabolisme : Phases

55
Métabolisme Réactions de phase I et II
Inactivation ou modification d’activité Augmentation de la polarité PHASE II Conjugaison: Formation d’un composé fortement polaire facilement éliminable Cycle enterohepatique

56
Réactions de phase I Oxydation : + O2 ou - H2 Réduction : + H2 ou - O2
Cytochrome P450 Réduction : + H2 ou - O2 Hydrolyse : + H2O BIOTRANSFORMATION

57
Réactions de phase II Conjugaison Meilleure solubilité (dans l’eau)
Acide glucuronique Sulfate Acétyl Meilleure solubilité (dans l’eau) Meilleure excrétion (ex: urine)
 Meilleure excrétion (ex: urine)")
58
Réactions de phase II Groupe transféré R’ Enzymes impliqués
Nom de la réaction Exemple Ac D-glucuronique UDP glucuronyl-transférase Glucuro-conjugaison Fénoprofène Sulfate Sulfotransférase Sulfo-conjugaison Paracétamol Méthyl Méthyltransférase Alcylation Captopril Glutathion Glutathion transférase Intoxication Paracétamol Acétyl Acétyl transférase Acétylation Isoniazide Glycocolle ou Glycine Glycyl-conjugaison Acide salicylique

59
Résultat de phase II Activation du pro-médicament
Inactif à actif Potentialisation: action supérieure Inactivation P:54

60
Métabolisme : Conséquences Pharmacologiques

61
Métabolisme : Conséquences Pharmacologiques

62
Métabolisme : Conséquences Pharmacologiques

63
Métabolisme :Rôle des enzymes

64
Le système monooxygénase à cytochrome P450
Métabolisme Le système monooxygénase à cytochrome P450 = système multienzymatique capable d’oxyder un composé exogène en lui transférant directement un atome d’oxygène à partir de l ’oxygène moléculaire de l’air.

65
Métabolisme

66
CYP450 = Une superfamille d’isoenzymes
Métabolisme CYP450 = Une superfamille d’isoenzymes

67
Métabolisme Réactions élémentaires catalysées par les cytochromes P450

68
Métabolisme

69
Biotransformation Inductif Inhibé Saturable Compétitif Variable
Individu Génétique Dose et voie d’administration

70
Biotransformation : facteurs de variation
1) Facteurs physico-chimiques : Structure chimique, Configuration spatiale, Liposolubilité. La forme galénique. 2) Facteurs pharmacodynamiques : Voie d’administration. Posologie.
 Facteurs physico-chimiques : Structure chimique, Configuration spatiale, Liposolubilité. La forme galénique. 2) Facteurs pharmacodynamiques : Voie d’administration. Posologie.")
71
Biotransformation : facteurs de variation
3) Facteurs physiologiques : La race, sexe, âge. Facteurs hormonaux, grossesse. Régime alimentaire. 4) Facteurs pathologiques : L’insuffisant hépatique, rénal. L’obésité, L’alcoolisme. Malnutrition. 5) Facteurs génétiques +++
 Facteurs physiologiques : La race, sexe, âge. Facteurs hormonaux, grossesse. Régime alimentaire. 4) Facteurs pathologiques : L’insuffisant hépatique, rénal. L’obésité, L’alcoolisme. Malnutrition. 5) Facteurs génétiques +++")
73
Métabolisme : induction /inhibition

74
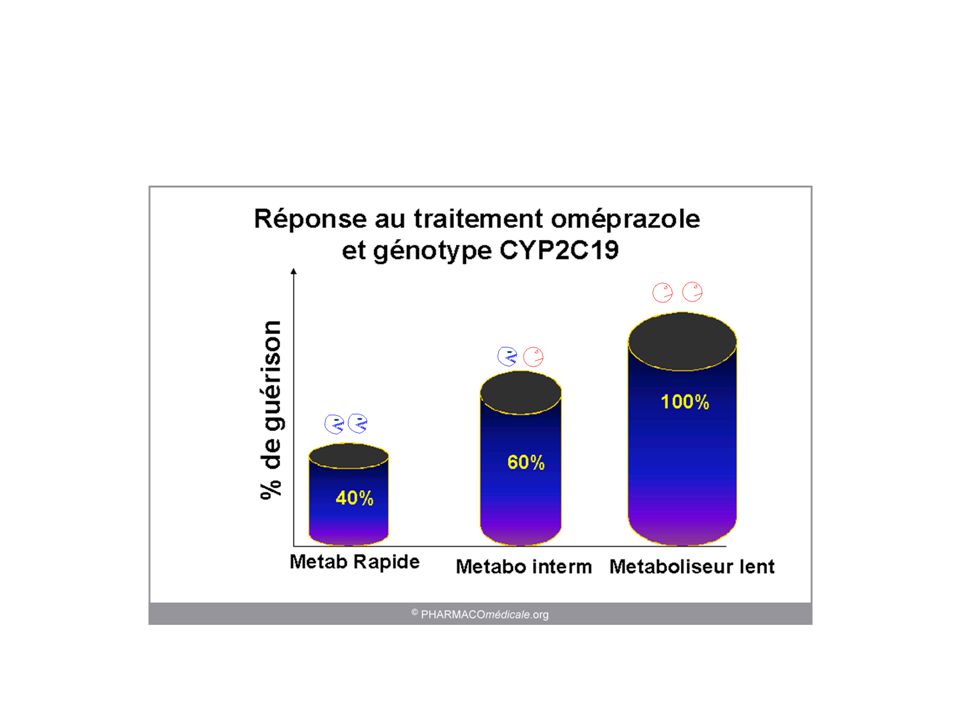
Métabolisme : facteurs de variation

75
Métabolisme : Paramètre de quantification
La clairance: capacité globale de l’organisme à éliminer une molécule = volume de plasma totalement épuré par unité de temps (ml/min) Clairance totale= Somme de clairance de chaque organe épurateur: foie, reins,intestins,poumons…. ClH = ClMH + ClB En pratique: on détermine à partir de prélèvements plasmatiques et urinaires - la clairance totale - la clairance rénale Facteurs de variations physio-pathologiques du métabolisme et de l’élimination = ajustement de la posologie et du rythme d’adm d’un traitement en fonction de la clairance.
 Clairance totale= Somme de clairance de chaque organe épurateur: foie, reins,intestins,poumons…. ClH = ClMH + ClB. En pratique: on détermine à partir de prélèvements plasmatiques et urinaires. - la clairance totale. - la clairance rénale. Facteurs de variations physio-pathologiques du métabolisme et de l’élimination = ajustement de la posologie et du rythme d’adm d’un traitement en fonction de la clairance.")
76
Paramètre de quantification
Métabolisme : Paramètre de quantification La clairance hépatique Clh = Qh x Eh débit sanguin hépatique mécanisme enzymatique (métabolisme) débit sanguin hépatique liaison aux protéines plasmatiques Diffusion Caff Débit (Q) E Cs EH = ( Ce – Cs ) / Ce varie de 0 à 1
 débit sanguin hépatique. liaison aux protéines plasmatiques. Diffusion. Caff. Débit (Q) E. Cs. EH = ( Ce – Cs ) / Ce. varie de 0 à 1.")
77
Métabolisme : Paramètre de quantification
Coefficient d’extraction fortement extraits E > 0,7 (phénotoine, diazépam). moyennement extraits 0,3 < E < 0,7 (codéine, aspirine). faiblement extraits E < 0,3 (morphine, propranolol).
. moyennement extraits 0,3 < E < 0,7 (codéine, aspirine). faiblement extraits E < 0,3. (morphine, propranolol).")
78
FOIE et MEDICAMENT Insuffisance hépatocellulaire : Biotransformations
Cholestase : Excrétion Biotransformations FOIE Biotransformation Excrétion

79
Modifications PK au cours d’IH
Augmentation de la biodisponibilité Ralentissement de l’absorption digestive Diminution de la fixation protéique Augmentation du volume de distribution Diminution des biotransformations

80
Ce qu’il faut retenir Le métabolisme : hépatique
But : Molécules hydrosolubles Acides fort Ionisés Conséquences pharmacologiques et toxicologiques

81
Pharmacocinétique Absorption Distribution Métabolisme Élimination

82
Élimination

83
Elimination : méthodes d’étude
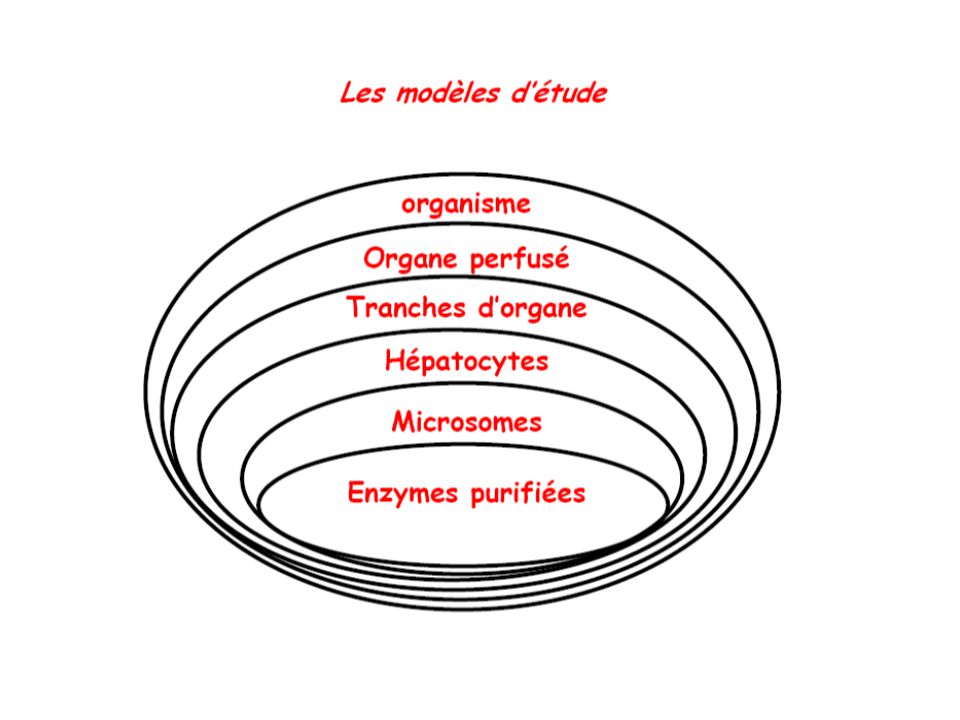
Méthode des clairances Microponction et microperfusion in vivo Tubule rénal isolé et perfusé in vitro Études biochimiques sur fragments de membrane

84
Elimination Élimination Directe Après métabolisme et/ou dégradation
Pulmonaire. Rénale. Après métabolisme et/ou dégradation Foie. Rein, poumon. Sang.

85
Élimination Elimination Elimination par le foie Elimination rénale
Excrétion par la bile Elimination par le tube digestif Réabsorption: cycle enterohépatique Elimination rénale Sous forme inchangée Sous forme dégradée: produits de dégradation Filtration de la forme non fixée Réabsorption possible de la forme non ionisée Les autres voies: salive, poumon, lait,..

86
Principe de l'excrétion rénale
Sang Réabsorption Filtrat Urine Sécrétion Absorption (eau et solutés) Sécrétion Filtration Eau et solutés
 Sécrétion. Filtration. Eau et solutés.")
87
Élimination Réabsorption (passive) Filtration Sécrétion (active)
Uniquement forme non ionisée, liposoluble. Essentiellement diffusion passive. Transport actif pour quelques composés: risque de compétition Importance du pH urinaire et des caractéristiques physico-chimiques du médicament. Mécanisme passif Molécules de PM < 68000 Seule la fraction libre est filtrée Réabsorption (passive) Filtration Tubule proximal Tubule collecteur Tubule distal Glomérule Sécrétion (active) Tube contourné proximal (TCP) Acides faibles / bases faibles Transport actif Saturable Phénomène de compétition Anse de Henle 87
 Filtration. Tubule proximal. Tubule collecteur. Tubule distal. Glomérule. Sécrétion. (active) Tube contourné proximal (TCP) Acides faibles / bases faibles. Transport actif. Saturable. Phénomène de compétition. Anse de Henle. 87.")
88
Paramètres de quantification ?

89
Élimination paramètres de quantification
La clairence rénale V excrétion rénale = V filtration + V sécrétion - V réabsorption V excrétion rénale C V filtration = V sécrétion - V réabsorption + CLR = CLfiltration + CLsécrétion - CLréabsorption 89 89

90
Demi-vie t½ est le temps nécessaire pour que la concentration plasmatique du médicament diminue de moitié Élimination ~~ totale : après 7 demi-vie

91
calcul de la demi-vie: C = C0 x e-Kt t1/2

92
Demi vie et équilibre Administration multiples
Conc. (mg) 5ème T1/2 T1/2= demi-vie Cmax Cmoy Cmin T1/2 Temps (h) Dose Dose Dose Dose Dose Dose Dose
 5ème T1/2. T1/2= demi-vie. Cmax. Cmoy. Cmin. T1/2. Temps (h) Dose. Dose. Dose. Dose. Dose. Dose. Dose.")
93
Demi-vie - quantification
Demi-vie courte: < 4h Demi-vie moyenne: entre 4 et 12h Demi-vie longue: > 12h

94
Demi-vie: ce qu’il faut retenir
Caractéristique d’un médicament Indépendante de la voie d’administration et de la dose Permet de prévoir l’état d’équilibre Permet le calcul de l’intervalle thérapeutique

95
ADME : cas particuliers
Nouveau-né et enfant Personne âgée Femme enceinte

96
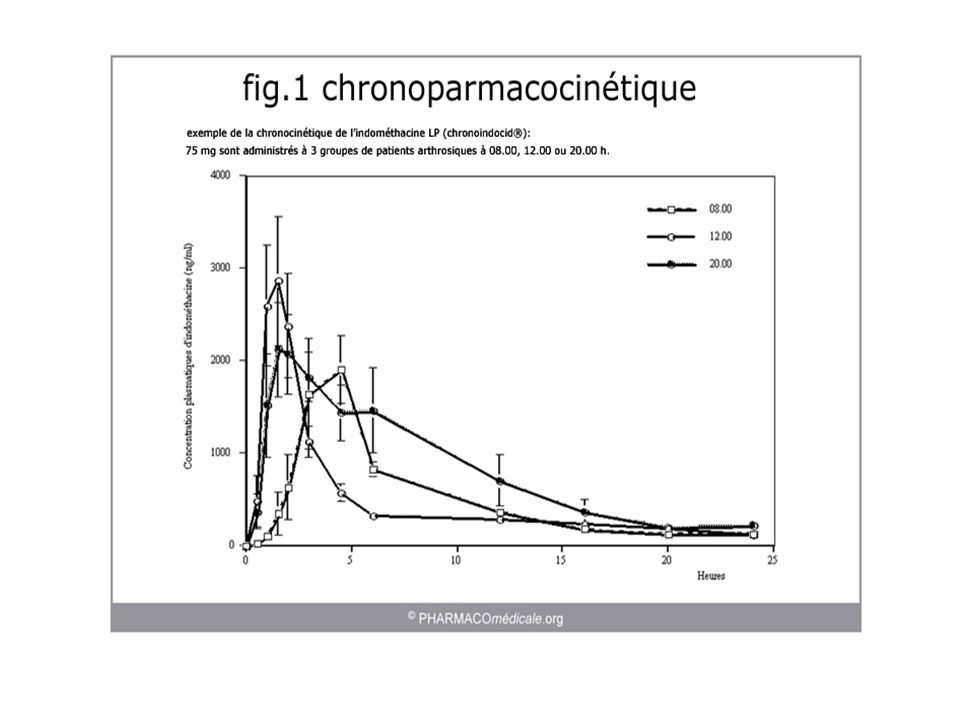
Chronopharmacologie Changements cycliques de ADME
Phénomènes biologiques de l’organisme sont rythmiques Conséquences : efficacité/tolérance en fonction du moment d’administration du médicament Pour agir un médicament doit se trouver à la bonne concentration au niveau de ses sites d'action... mais également au bon moment.

98
Surveillance thérapeutique
Mesure de la concentration sérique d’un médicament Fenêtre thérapeutique étroite Concentration minimale efficace Concentration minimale toxique CME 10mg/l CTM 20 mg/l Fenêtre thérapeutique

99
A retenir Pharmacocinétique: étude du devenir du médicament dans l’organisme dans le but Adapter: posologie voie d’administration rythme d’administration d’un médicament pour un patient donné => Optimisation de l’efficacité du médicament


















cinétique/pharmaco (toxico)dynamie>")





