Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
M. Pourrat Service Pharmacie Hôpital Beaujon
Pharmacocinétique. M. Pourrat Service Pharmacie Hôpital Beaujon

2
Introduction Pharmacocinétique:
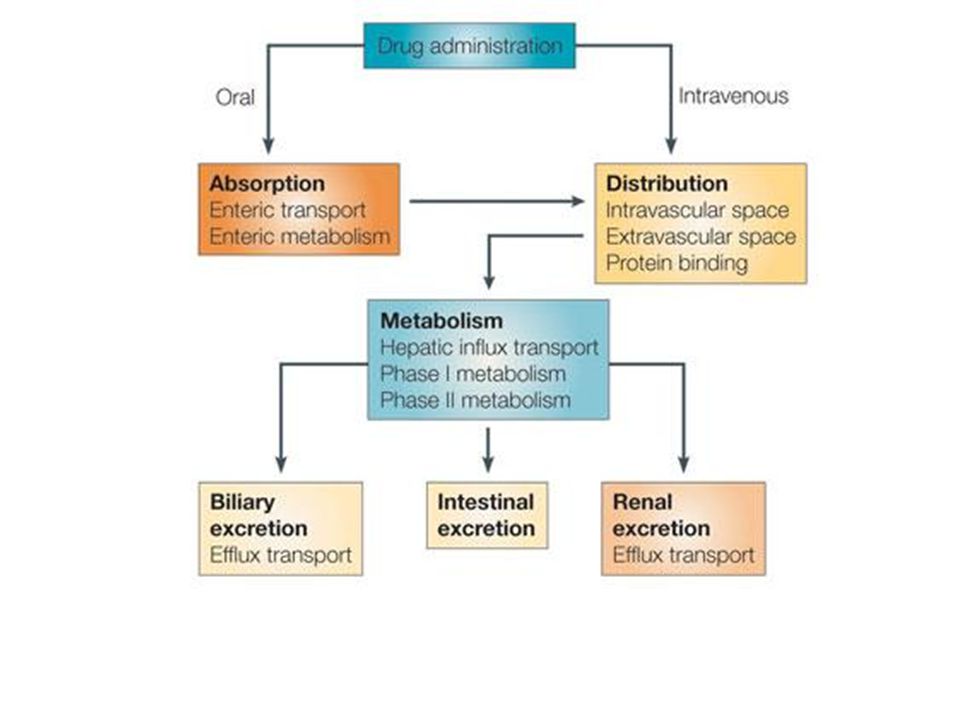
Étude du devenir d’un principe actif (PA) contenu dans un médicament dans l’organisme. 4 phases: Absorption Distribution Métabolisme Elimination
 contenu dans un médicament dans l’organisme. 4 phases: Absorption. Distribution. Métabolisme. Elimination.")
3
Détermination des paramètres pharmacocinétique d’un PA apporte des informations permettant de choisir les voies d’administration, la forme galénique, adapter les posologies.

4
Plan I - Pharmacocinétique descriptive: ADME. 1. Absorption
2. Distribution 3. Métabolisme 4. Élimination II – Influence physiologiques et physiopathologiques sur la pharmacocinétique des médicaments. III - Pharmacocinétique analytique. 1. Influence de la dose 2. Influence de la fréquence d’administration

5
I - Pharmacocinétique descriptive: ADME.
1 – Absorption PA doit traverser les membranes biologiques du site d’absorption vers le sang pour pénétrer dans la circulation générale. Doit franchir des obstacles: Voie orale: paroi du TD, Voie cutanée ou transdermique: peau (pommade, patch), Voie rectale: paroi du rectum (suppo)…
, Voie rectale: paroi du rectum (suppo)…")
6
Appareil digestif

7
Conditions d’absorption relatives au mdc:
Hydrosolubilité / liposolubilité, forme neutre: mieux absorbée taille de la molécule Forme galénique Condition d’absorption liées au milieu: Estomac: très acide => acide faible mieux absorbé Vidange gastrique, mobilité intestinale, L’alimentation Prise simultanée de Mdc L’âge Pathologies associées

9
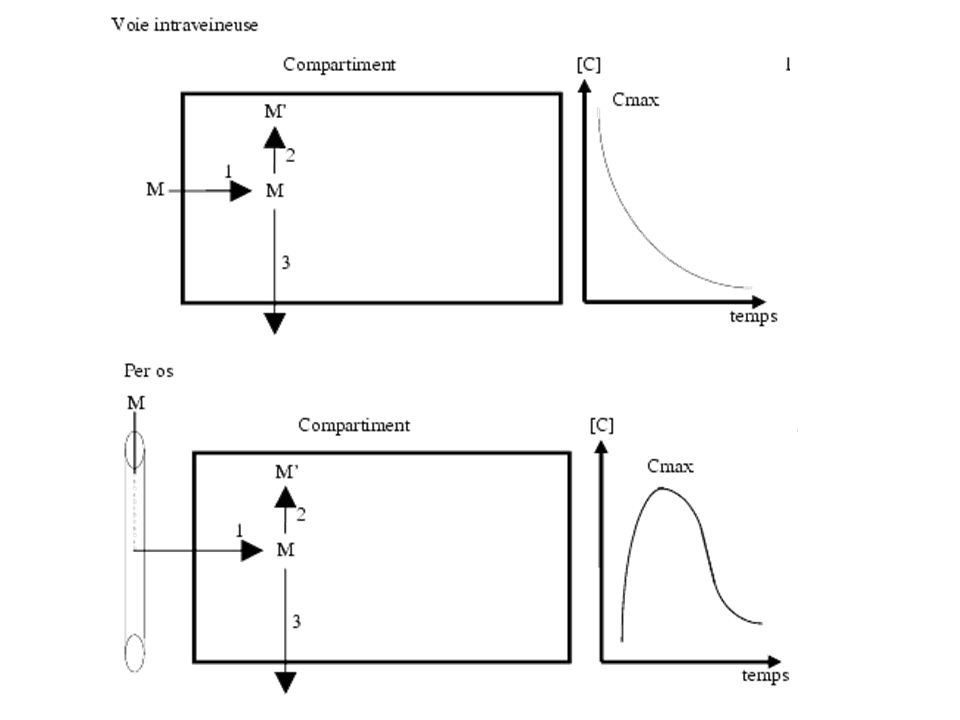
Notion de premier passage (schéma).
Effet de premier passage évité pour les suppo. Biodisponibilité: fraction de la dose de Mdc qui atteint la circulation générale et la vitesse à laquelle elle l’atteint (Cmax et Tmax). => Voies IV+++, sublinguale, transdermique.
. => Voies IV+++, sublinguale, transdermique.")
11
Biodisponibilité: notion indépendante de l’efficacité! Métabolites actifs prodrogues

14
Seul le Mdc libre est actif.
2 – Distribution Liaison du Mdc plus ou moins aux protéines du plasma (albumine) selon nature du PA: Liaison forte: phénytoïne. Liaison moyenne: théophylline. Liaison faible: paracétamol. Seul le Mdc libre est actif.
 selon nature du PA: Liaison forte: phénytoïne. Liaison moyenne: théophylline. Liaison faible: paracétamol. Seul le Mdc libre est actif.")
15
Accumulation plus ou mois dans certains organes selon affinité pour
Graisses Tissus cibles… Passage dans le Système nerveux central et le liquide céphalo-rachidien: passivement: molécules lipophiles, gaz anesthésiques.. Par un transporteur: L-dopa… Quantifié par le volume de distribution.

16
Diffusion tissulaire. Distribution dans espace extra-cellulaire mais aussi intracellulaire. Même problème que pour l’absorption digestive. Volume apparent de distribution. V = dose / Co V = dose / (AUC x k) k = constante d’élimination.
 k = constante d’élimination.")
17
Facteurs modifiant la distribution
Volume liquidien de l’organisme: age, deshydratation. Rapport masse maigre/tissu adipeux: obésité, age. Hémodynamique: état de choc, insuffisance cardiaque chronique. Modification des protéines plasmatiques: diminution de l’albumine+++ grossesse, dénutrition, grands brûlés, cirrhose, état inflammatoire, état infectieux.

18
Réactions de phase I: oxydation, réduction, hydrolyse.
3 – Métabolisme. Transformation du Mdc par le système enzymatique, surtout au niveau du foie. Réactions de phase I: oxydation, réduction, hydrolyse. Réactions de phase II: conjugaison (ac gluc) Plusieurs réactions possibles pour un Mdc Certains Mdc modifient l’activité des enzymes (inducteurs, inhibiteurs) => interaction médicamenteuse+++ Métabolites: inactifs, actifs, activité toxique.
 Plusieurs réactions possibles pour un Mdc. Certains Mdc modifient l’activité des enzymes (inducteurs, inhibiteurs) => interaction médicamenteuse+++ Métabolites: inactifs, actifs, activité toxique.")
19
Différences liées au malade:
Génétique: métaboliseurs lents, intermédiaires, rapides.+++ Physiologique: métabolisation parfois plus lente chez le nouveau-né que chez l’adulte. Pathologique

20
PA tend à être évacué par
4 – Élimination PA tend à être évacué par Excrétion (rein) Métabolisation (foie). Parfois cycle entéro-hépatique Autres: salive, poumons, lait! Elimination sous forme inchangée et/ou sous forme de un/plusieurs métabolites. Quantification = clairance. => Volume sanguin ou plasmatique totalement débarrassé de la substance par unité de temps (débit).
 Métabolisation (foie). Parfois cycle entéro-hépatique. Autres: salive, poumons, lait! Elimination sous forme inchangée et/ou sous forme de un/plusieurs métabolites. Quantification = clairance. => Volume sanguin ou plasmatique totalement débarrassé de la substance par unité de temps (débit).")
21
Demi-vie d’un médicament.
T1/2 = temps nécessaire pour passer d’une concentration plasmatique à sa moitiée, quel que soit cette concentration. Détermination du rythme posologique. Estimation du temps mis pou atteindre le plateau d’équilibre = 5 x T1/2

22
II – Influence physiologiques et physiopathologiques sur la pharmacocinétique des médicaments.
Selon la pharmacocinétique du Mdc. Insuffisance rénale Diminuer la dose Augmenter l’intervalle d’administration de la même dose Insuffisance hépatique Difficile à appréhender: perturbations de la synthèse des protéines, de l’excrétion biliaire.

23
III - Pharmacocinétique analytique.
1 – Influence de la dose. Effet pharmacologique d’un PA lié à la concentration plasmatique. Seuil thérapeutique: concentration minimale au dessous de laquelle aucune activité n’est obtenue. Limite supérieure: concentration maximale au-delà de laquelle apparaissent les effets indésirables Intervalle thérapeutique: zone intermédiaire dans laquelle les concentrations sont à la fois active et non-toxiques. => Index thérapeutique.

24
C Effets indésirables Intervalle thérapeutique Absence d’effet pharmacologique T

25
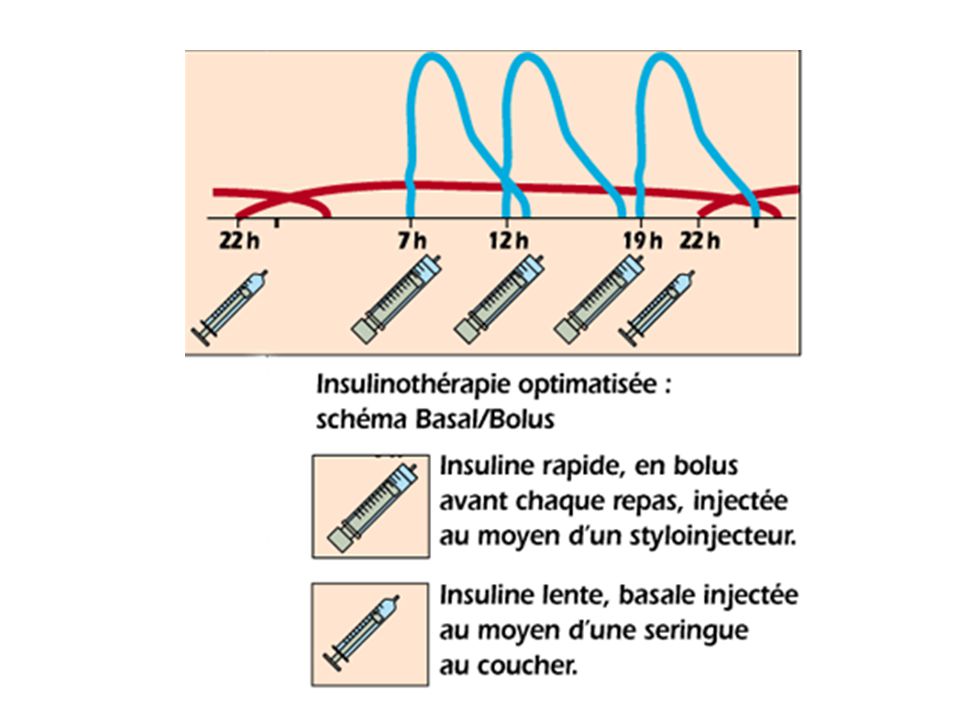
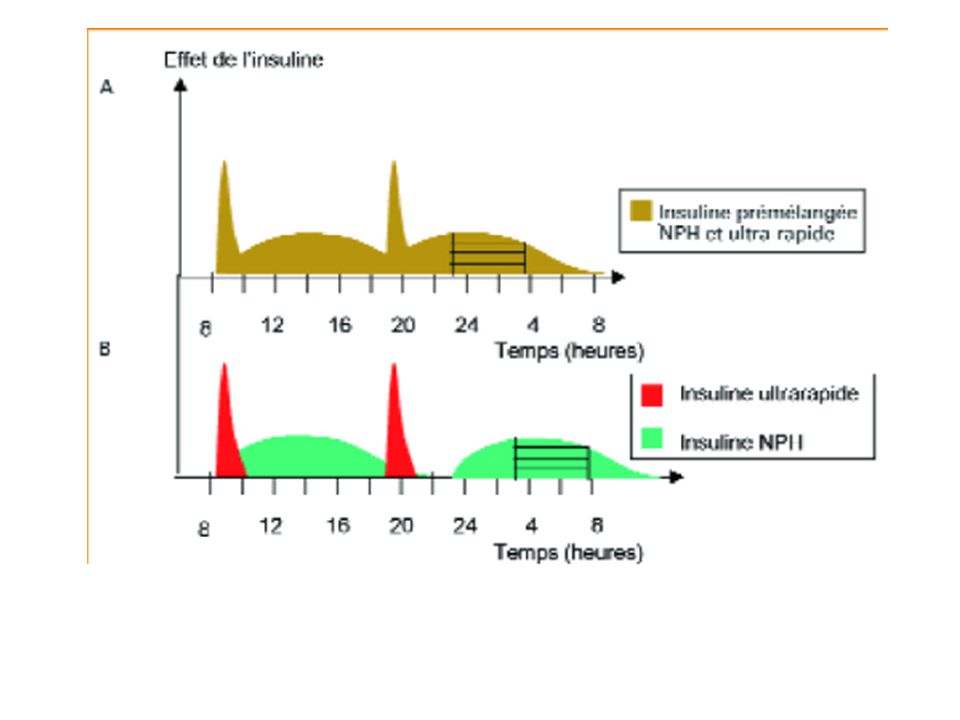
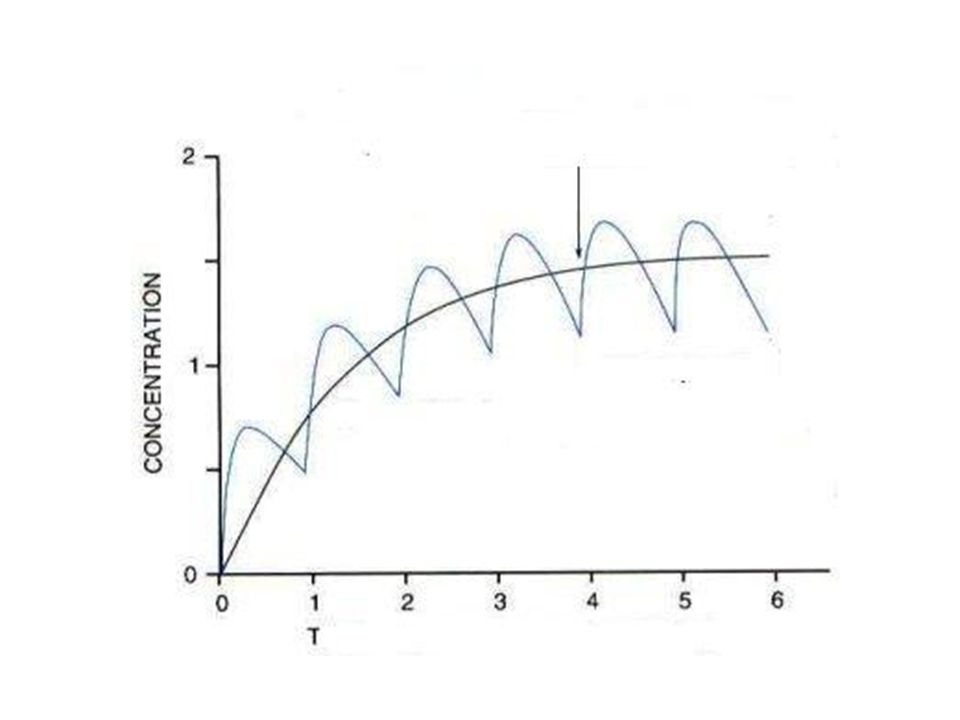
2. Influence de la fréquence d’administration.
Administration réitérée des Mdc. Objectif: Obtenir une efficacité thérapeutique Maintenir en permanence une concentration plasmatique active dans l’intervalle thérapeutique Éviter tout phénomène d’accumulation pouvant conduire à l’apparition d’effets toxiques.

27
CONCLUSION Interaction médicamenteuses de type pharmacocinétique: tout au long du processus ADME. Surtout au niveau des enzymes (Cytochrome P450). Dosage des médicaments.

Présentations similaires













cinétique/pharmaco (toxico)dynamie>")






