Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
LE CAS DE LA PETITE MARGAUX
Néonatologie, le

2
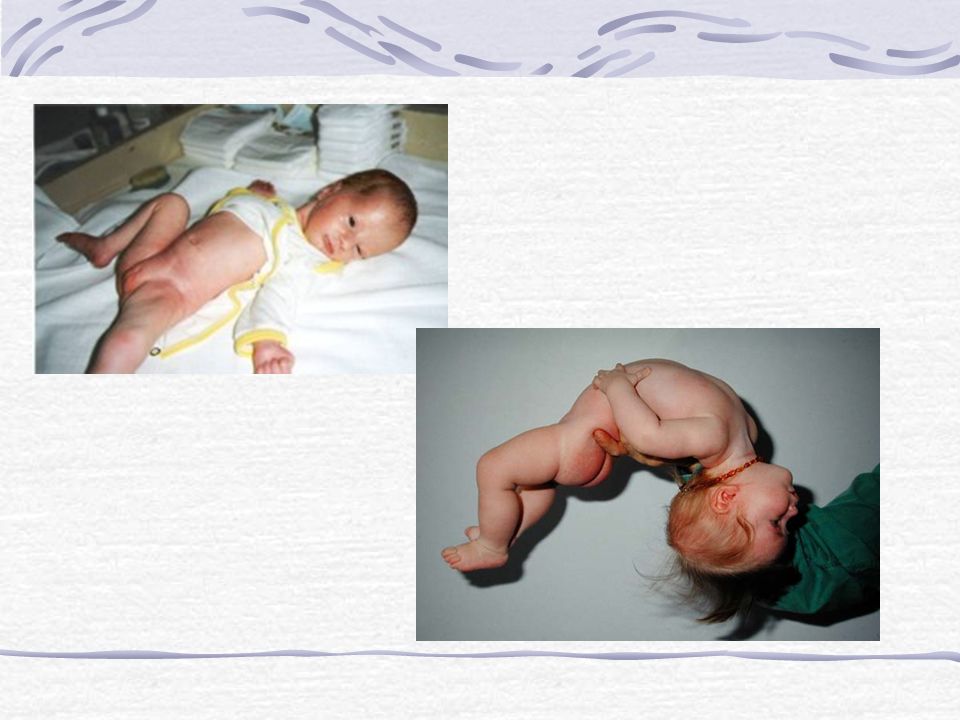
Margaux est née le Transfert d’Epernay à J0 pour hypotonie globale et mvse adaptation néonatale ATCD : - Mère née le - G1P1, Gpe A+ - Asthme, aide-soignante - Père: jumeau homozygote, ouvrier viticole, souffle mitral

3
Grossesse: - Suivi obstétrical sur Epernay - Terme le - Sérologies: Toxo +, Rub +, HBV-, HIV -, HCV-, SYPH - - ECBV négatif, RAI négative - ATCD: métrorragie 1er trim, glycosuries - - HT21: 1/1166 Accouchement Epernay le - Césarienne programmée pour siège et bassin étroit à 39SA - LA clair - Sous Rachianesthésie, extraction longue et difficile - Vomissement maternel pendant l’extraction

4
Etat à la naissance: - Terme: 39 SA - Apgar: 3.8.8 - PN: 2825g T: 50cm PC: 34.5cm - Extraction difficile - Ventilation par la SF, somnolence - Mis sous 3l/min O2

5
- Dextro: 0.41 g/L resucrée avec des dextro restant bas
- Semble algique au niv occipitale dc mise sous Efferalgan - Reste somnolente, pas de cri mais eupnéique - Pas de succion-déglutition - Transfert URIP

6
A l’URIP Examen identique Alim: Régime enrichi n°1 pompe 1h/3
Qq bradycardies et désaturations. Normalisation des dextros (limite inférieure)
")
7
HYPOTHESES - SFA +++ - IMF - Métabolique - Neurogéniques
- Anomalies chromosomiques

8
Bilan Biologique: - Gpe A + coombs - - Periph stériles, HC stérile, CRP - - GDS N - Iono N, Bili N, ASAT 2N (hémolyse), ALAT N - Calcémie N - Ac lactique 2.1 mmol/l N - Ammoniémie: 33 µmol/l N - CAA et CAO en cours

9
ETF: normale EEGa: pas de décrochage, modulation positive EEG: lent, discontinu, sans crise 2e EEG : idem Rx Crâne N Echocardiographie N Conclusion: somnolence et hypotonie sur SFA modérée

10
EN NEONATOLOGIE Reste hypotonique et difficultés alimentaires
RGO: Primperan et Gumilk Echo de hanches: laxité et instabilité hanche droite et hanche gauche luxable mais réductible Avis ortho: coussin d’abduction et suivi

11
EN NEONATOLOGIE Echo vésicorénale normale
1ère CAO: Hyperlactacidurie et Augmentation des ac Dicarboxyliques 2è CAO: Hyperlactacidurie, discussion cycle oxydo-redox CAA: normale Ac pyruvique et lactique sériques Nx

14
RESULTATS Caryotype: 46 XX
FISH: délétion interstitielle du chrms 15 en position q11-q13 (locus du gène SNRPN)
")
15
SYNDROME DE PRADER-WILLI
CONCLUSION SYNDROME DE PRADER-WILLI

16
En y regardant de plus près…
Argument en faveur: - Hypotonie néonatale +++ - Hypoplasie des grandes lèvres - Dysmorphie faciale - Troubles alimentaires

17
En y regardant de plus près…
Arguments en défaveur: - Augm des ac. dicarboxyliques - Hyperlactacidurie

18
Qu’est ce que le SPW ? Décrit en 1956 par les Dr Prader, Willi et Labhart Mie génétique rare svt accidentelle Cas familiaux rares Anomalie du chromosome 15 Affection très hétérogène

19
Qu’est ce que le SPW ? Définition:
- Dysfonctionnement hypothalamo-hypophysaire - Associé à une hypotonie majeure en période néonatale et les 2 premières années de vie - Puis se complique avec l’âge de troubles alimentaires (hyperphagie et risque d’obésité morbide), du comportement, de l’apprentissage et parfois de troubles psychiatriques
, du comportement, de l’apprentissage et parfois de troubles psychiatriques.")
20
Qu’est ce que le SPW ? Epidémiologie:
- Une des causes les plus fréquentes d’obésité génétique - Incidence: 1/25000

21
CLINIQUE Période néonatale:
- Hypotonie (axiale) sévère justifiant svt une hospit prolongée en néonat - Tbles de la succion-déglutition - Difficulté de prise pondérale - Dysmorphie faciale discret mais constant - Extrémités petites
 sévère justifiant svt une hospit prolongée en néonat. - Tbles de la succion-déglutition. - Difficulté de prise pondérale. - Dysmorphie faciale discret mais constant. - Extrémités petites.")
23
CLINIQUE Dans la petite enfance:
- Amélioration partielle de l’hypotonie mais qui persistera toute la vie - Retard à la marche (moyenne 24 mois) - Dysarthrie - Difficultés masticatoires - Faiblesses des muscles respiratoires: infections respi récidivantes
 - Dysarthrie. - Difficultés masticatoires. - Faiblesses des muscles respiratoires: infections respi récidivantes.")
24
CLINIQUE A partir de 2 ans:
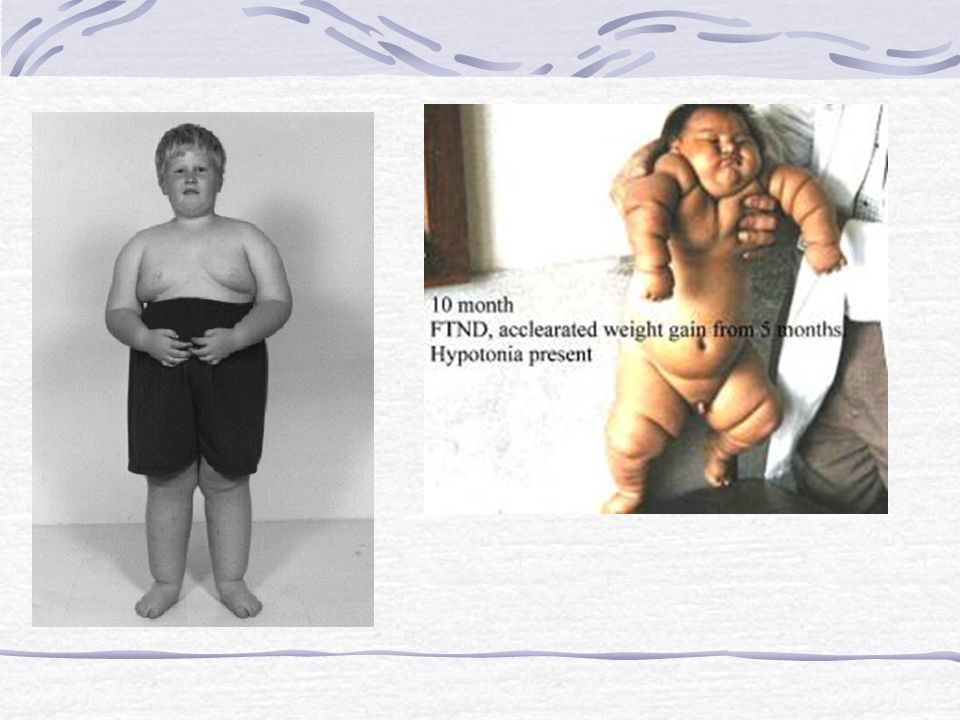
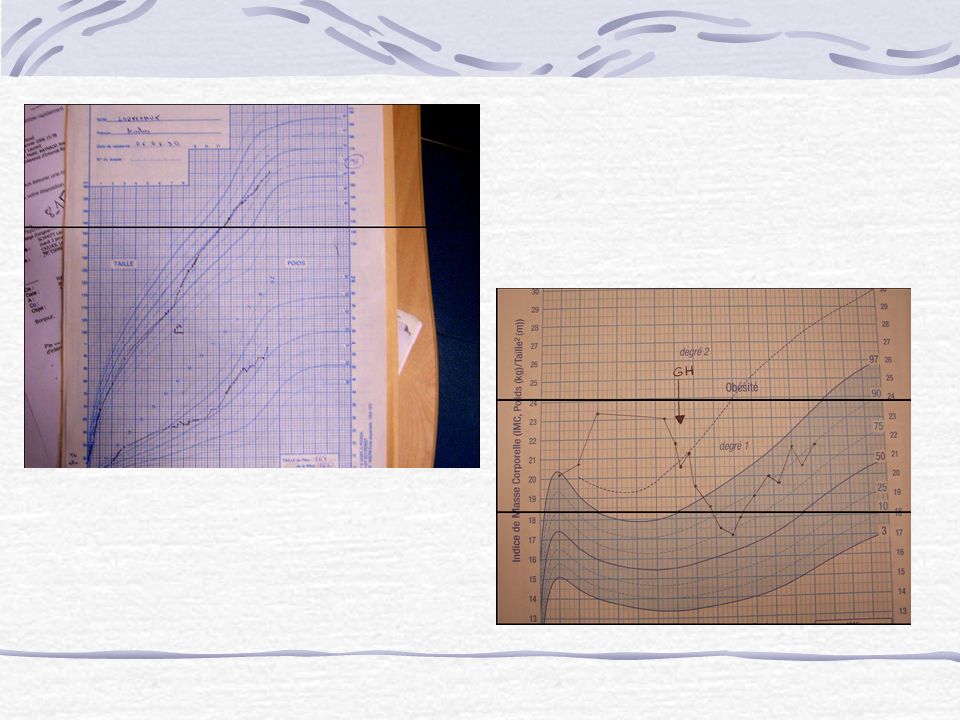
- Constitution d’une obésité +++ (hyperphagie et baisses des dépenses énergétique de base) Défaut de régulation de la satiété (non connu) - Retard statural par déficit en GH (50-100%) - Puberté incomplète par hypogonadisme partiel: * micropénis et cryptorchidie * développement mammaire et menstruation retardés et irréguliers
 Défaut de régulation de la satiété (non connu) - Retard statural par déficit en GH (50-100%) - Puberté incomplète par hypogonadisme partiel: * micropénis et cryptorchidie. * développement mammaire et menstruation retardés et irréguliers.")
26
CLINIQUE Sur le plan neurodéveloppementale:
- Retard mental rarement majeur, très variable - Difficulté d’apprentissage, tble oral

27
CLINIQUE Sur le plan psychocomportemental:
- névroses et psychoses rare chez l’enfant - Evocateur: crise brutale de colère incontrôlable, lors de frustration mineures, parfois violence physique, comportement obsessionnel, répétitions verbales, tendance dépressive

28
CLINIQUE Complications liées à l’hypotonie: - Fatigabilité à l ’effort
- Maladresse - Trouble de l’élocution - Orthopédiques: LCH, scoliose - Ophtalmo: strabisme, myopie, astigmatie, hypermétropie

29
CLINIQUE Autres complications:
- Dermato: prurit (6 ans) asso baisse de sensibilité à la douleur - Dysrégulation thermique - Neuro: épisodes convulsifs - Tble du sommeil, endormissement diurne - SAOS même sans obésité - Dentaire: carie, ano émail (salive épaisse)
 asso baisse de sensibilité à la douleur. - Dysrégulation thermique. - Neuro: épisodes convulsifs. - Tble du sommeil, endormissement diurne. - SAOS même sans obésité. - Dentaire: carie, ano émail (salive épaisse)")
30
CLINIQUE Autres complications:
- Digestives: fragilité intestinale, difficultés de vomissement (tonus vagal déficient) - Hémato: fragilité vasculaire et hématomes
 - Hémato: fragilité vasculaire et hématomes.")
31
MORBIMORTALITE Conditionné par l’obésité: - Diabète
- Complications cardiorespiratoires - Lymphoedème et troubles trophiques - Ménopause précoce et csq * A noter: de très rares cas de grossesses ont été rapportés

32
ETIOPATHOGENIE Etiologie génétique suspectée dès 1956 mais démontrée qu’en 1985 Délétion de petite taille du chromosome 15 d’origine paternelle dans la région 15q11-q13

33
ETIOPATHOGENIE Trois mécanismes:
- Délétions interstitielles (70%), plus rarement translocation déséquilibré (5%) svt de novo - Disomie uniparentale maternelle (25%) - Microdélétion indétectable (1-2%): mutation d’empreinte
, plus rarement translocation déséquilibré (5%) svt de novo. - Disomie uniparentale maternelle (25%) - Microdélétion indétectable (1-2%): mutation d’empreinte.")
34
ETIOPATHOGENIE Anomalies accidentelles et sporadiques
Pas de risque de récurrence lors prochaines grossesses Sauf mutation empreinte et translocation transmise par le père Cas familiaux rares

35
ETIOPATHOGENIE Gènes impliqués dans le SPW = 2Mb soit 50% de la région 15q11-q13 3 sites de cassures: BP1, BP2 et BP3 Pls gènes: SNRPN, Necdin, Magel 2, snoARN Hétérogénéité phénotypique: délétions plus typiques que les disomies

36
PHYSIOPATHOLOGIE Lien avec un dérèglement de la fonction hypothalamique Mais pas d’ano majeurs sur l’imagerie sauf lègère hypotrophie antéhypophysaire (IRM) Pas de déficit ni de résistance à la leptine Hyperghrelinémie produite par l’estomac et qui stimule l’appétit (inverse pour l’obésité commune) Lien ghreline et GH, sommeil, mémoire…
 Pas de déficit ni de résistance à la leptine. Hyperghrelinémie produite par l’estomac et qui stimule l’appétit (inverse pour l’obésité commune) Lien ghreline et GH, sommeil, mémoire…")
37
DIAGNOSTIC Critères majeurs (valeur : 1 point chacun)
Les critères de diagnostic du syndrome de Prader-Willi présentés ci-dessous sont inspirés de Holm et al. (Pediatrics 91, 398, 1993) Critères majeurs (valeur : 1 point chacun) 1. Hypotonie centrale entraînant une difficulté à téter, s'améliorant progressivement avec l'âge. 2. Chez le NRS, difficulté à s'alimenter, recours au gavage, mvse prise de pds 3. Prise de pds excessive entre 1 et 6 ans +/- obésité centrale
 Critères majeurs (valeur : 1 point chacun) 1. Hypotonie centrale entraînant une difficulté à téter, s améliorant progressivement avec l âge. 2. Chez le NRS, difficulté à s alimenter, recours au gavage, mvse prise de pds. 3. Prise de pds excessive entre 1 et 6 ans +/- obésité centrale.")
38
DIAGNOSTIC Critères majeurs (suite):
4. Visage caractéristique ; dolichocéphalie chez le NRS, visage ou diamètre bifrontal étroits, yeux en amande ; la bouche paraît petite, lèvre sup mince, les commissures tombantes. (mini 3 nécessaires). 5. Hypogonadisme, corresp à l'une des caract. selon l'âge : • Hypoplasie génitale (hypoplasie scrotale, cryptorchidie, pénis et/ou testicules de petite taille, absence ou hypoplasie sévère des petites lèvres et/ou du clitoris) • Maturation gonadale tardive ou incomplète ; apparition tardive des signes de puberté après 16 ans.
. 5. Hypogonadisme, corresp à l une des caract. selon l âge : • Hypoplasie génitale (hypoplasie scrotale, cryptorchidie, pénis et/ou testicules de petite taille, absence ou hypoplasie sévère des petites lèvres et/ou du clitoris) • Maturation gonadale tardive ou incomplète ; apparition tardive des signes de puberté après 16 ans.")
39
DIAGNOSTIC Critères majeurs (suite):
6. Retard global du dvt avant 6 ans; chez les enfants + âgés, difficultés d'apprentissage ou RM léger à modéré. 7. Hyperphagie (appétit excessif): recherche constante, obsession 8. Délétion 15q11-13 (>650 bandes, confirmée de préférence par le test FISH ) ou autre ano moléculaire appropriée dans cette région du chrms, notamment une disomie maternelle.
: recherche constante, obsession. 8. Délétion 15q11-13 (>650 bandes, confirmée de préférence par le test FISH ) ou autre ano moléculaire appropriée dans cette région du chrms, notamment une disomie maternelle.")
40
DIAGNOSTIC Critères mineurs (0.5 pts):
1. Diminution des MAF ou léthargie infantile ou faiblesse du cri chez le NRS, s'améliorant avec l'âge. 2. Pb de comportement caract. : crises de colère, accès de violence, persévération, comportement obsessif/compulsif, tendance à ergoter, à faire de l'opposition systématiq, à se montrer rigide, manipulateur, possessif et entêté, à voler, à mentir. (mini 5 nécess) 3. Trouble du sommeil ou apnée nocturne. 4. Petite taille à l'âge de 15 ans, comparée aux autres membres de la famille (en l'absence de ttt par GH).
 3. Trouble du sommeil ou apnée nocturne. 4. Petite taille à l âge de 15 ans, comparée aux autres membres de la famille (en l absence de ttt par GH).")
41
DIAGNOSTIC Critère mineurs (suite):
5. Hypopigmentation : teint et cheveux clairs comparés à ceux des autres membres de la famille. 6. Mains petites (<25e p ) et/ou pieds petits (<10e p) pr la taille 7.Mains étroites à la tranche droite. 8. Anomalies oculaires (myopie, ésotropie). 9. Salive épaisse et visqueuse ayant tendance à sécher aux commissures des lèvres. 10. Langage : problèmes d'articulation. 11. Tendance à se gratter.
 et/ou pieds petits (<10e p) pr la taille. 7.Mains étroites à la tranche droite. 8. Anomalies oculaires (myopie, ésotropie). 9. Salive épaisse et visqueuse ayant tendance à sécher aux commissures des lèvres. 10. Langage : problèmes d articulation. 11. Tendance à se gratter.")
42
DIAGNOSTIC Autres indices:
1. Seuil de tolérance à la douleur élevé. 2. Moins de vomissements que la normale. 3. NRS : instabilité de la T°c corporelle. Enfants + âgés ou adultes : sensibilité altérée de la thermorégulation. 4. Scoliose ou cyphose. 5. Développement précoce de la pilosité pubienne ou axillaire (avant 6 ans) 6. Ostéoporose (déminéralisation ou amincissement des os). 7. Capacité inhabituelle à assembler des puzzles. 8. Investigations neuromusculaires normales
 6. Ostéoporose (déminéralisation ou amincissement des os). 7. Capacité inhabituelle à assembler des puzzles. 8. Investigations neuromusculaires normales.")
43
DIAGNOSTIC Score avant 3 ans: 5 pts dont 4 majeurs
Score après 3 ans: 8 pts dont 5 majeurs

44
DIAGNOSTIC Etude cytogénétique par FISH ou l’étude en haute résolution de la bande 650 permet le diagnostic dans 70 % des cas

45
DIAGNOSTIC Biologie Moléculaire: étude de la méthylation de la région 15q proximale

46
DIAGNOSTIC DIFFERENTIEL
L’hypotonie néonat est aussi présente ds : Anomalie chromosomique (dup Xq27.2-ter, del 6q16.2, del 1p36, del 10q26) Syndrome de l'X fragile Syndrome de Rett Syndrome d'Angelman L’association retard de développement, obésité et hypogonadisme se rencontre dans : Syndrome de Borjeson-Forssman-Lehmann Syndrome de Bardet-Biedl Syndrome de Cohen
 Syndrome de l X fragile. Syndrome de Rett. Syndrome d Angelman. L’association retard de développement, obésité et hypogonadisme se rencontre dans : Syndrome de Borjeson-Forssman-Lehmann. Syndrome de Bardet-Biedl. Syndrome de Cohen.")
47
ANNONCE DU DG Meilleurs chances d’intégration si dg posé en période néonatale Se montrer optimiste L’obésité n’est pas inéluctable Préparer aux tbles apprentissage Préparer au suivi psychologique Insister sur le rôle des parents et les soutenir

48
PRISE EN CHARGE De 0 à 2 ans: pluridisciplinaire
- Gavage, alimentation prudente, éducation à l’alimentation - Ne pas sous-alimenter +++ - Kiné mobilisatrice 2/sem - Psychomotricité (CAMSP, CMPP) - Orthophonie (instrumentation, communication verbale) - Prise en charge 100 %, aides admi (AES…) - Endocrino: GH dès 6 mois, thyro +/-
 - Orthophonie (instrumentation, communication verbale) - Prise en charge 100 %, aides admi (AES…) - Endocrino: GH dès 6 mois, thyro +/-")
50
PRISE EN CHARGE De 2 à 12 ans: Hyperphagie +++
- Régime alimentaire adapté permanent - Soins dentaires - Orthophonie - Ttt lésion cutanées - Ttt complications orthopédiques - Suivi ophtalmo - Psychomotricité - Activité physique

51
PRISE EN CHARGE De 2 à 12 ANS: - Chir abaissement testiculaire
- Scolarisation: normale le plus lgtps possible - Suivi psychologique voire psychiatrique - Endocrinologique +++: GH et suivi - Rôle des CAMSP, CMPP… - Aides administratives +++

52
PRISE EN CHARGE A l’adolescence: - Endocrino: GH et puberté +/- THS
- Nutrition +++ - Suivi psy +++: psychoth, soutien fam

53
CONSEIL GENETIQUE Majorité des cas: sporadique
Si microdéletion interstitielle paternelle ou disomie mat de novo: rassurant mais dg prénatal peut être proposé du fait du risque de mosaïcisme germinal (risque de FCS) Risque élevée si translocation équilibré, ano d’empreinte: Enquête familiale et dg prénatal +++
 Risque élevée si translocation équilibré, ano d’empreinte: Enquête familiale et dg prénatal +++")
54
EVOLUTION 3 mois Dernier poids: 3820 g (2mois ½) Hypotonie persistante
Boit bien Dépistage génétique des 2 parents

Présentations similaires





>")




>")



 Pr E. Tournier-Lasserve>")






