Télécharger la présentation
1
Recherche de motifs par méthodes exploratoires: Comparaisons de performances et statistiques sur le score

2
Définitions: Un motif est un ensemble doccurrences dans les séquences (taille contrainte) La répartition des occurrences est contrainte à une par séquence. La fonction objectif est lentropie relative (ou ratio des vraisemblances)
.")
3
Définitions P: espace des motifs (~lmoy N ) M: espace des mots (K W ) Deux opérateurs permettent détablir une correspondance entre les deux espaces.
 M: espace des mots (K W ) Deux opérateurs permettent détablir une correspondance entre les deux espaces.")
4
Définitions Q: espace des motifs représentables par un mot (selon lopérateur de projection) Q est inclu dans P Q a au plus la taille de M M P Q
 Q est inclu dans P Q a au plus la taille de M M P Q")
5
Les déplacements dans P (hill climbing) 1) Voisinage par séquence (Lawrence et al., Science, 1993) Un point de P (un motif) est représenté par un vecteur dentiers à N dimensions, chaque dimension représentant une position sur la séquence correspondante. On ne modifie quune dimension à la fois. Les dimensions sont prises dans un ordre prédéfini, ou aléatoirement, sans remise. On choisit la position qui maximise la fonction objectif (entropie relative)
.")
6
? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? Les déplacements dans P (hill climbing) 1) Voisinage par séquence (Lawrence et al., Science, 1993) Un point de P (un motif) est représenté par un vecteur dentiers à N dimensions, chaque dimension représentant une position sur la séquence correspondante. On ne modifie quune dimension à la fois. Les dimensions sont prises dans un ordre prédéfini, ou aléatoirement, sans remise. On choisit la position qui maximise la fonction objectif (entropie relative)
.")
7
Les déplacements dans P (hill climbing) 1) Voisinage par séquence (Lawrence et al., Science, 1993) Un point de P (un motif) est représenté par un vecteur dentiers à N dimensions, chaque dimension représentant une position sur la séquence correspondante. On ne modifie quune dimension à la fois. Les dimensions sont prises dans un ordre prédéfini, ou aléatoirement, sans remise. On choisit la position qui maximise la fonction objectif (entropie relative)
.")
8
Les déplacements dans P (hill climbing) 2) Phase shift (Lawrence et al., Science, 1993) Le voisinage comprend lensemble des vecteurs de position relatifs. Opérateur destiné à corriger les motifs mal calés

9
Les déplacements dans P (hill climbing) 2) Phase shift (Lawrence et al., Science, 1993) Le voisinage comprend lensemble des vecteurs de position relatifs. Opérateur destiné à corriger les motifs mal calés

10
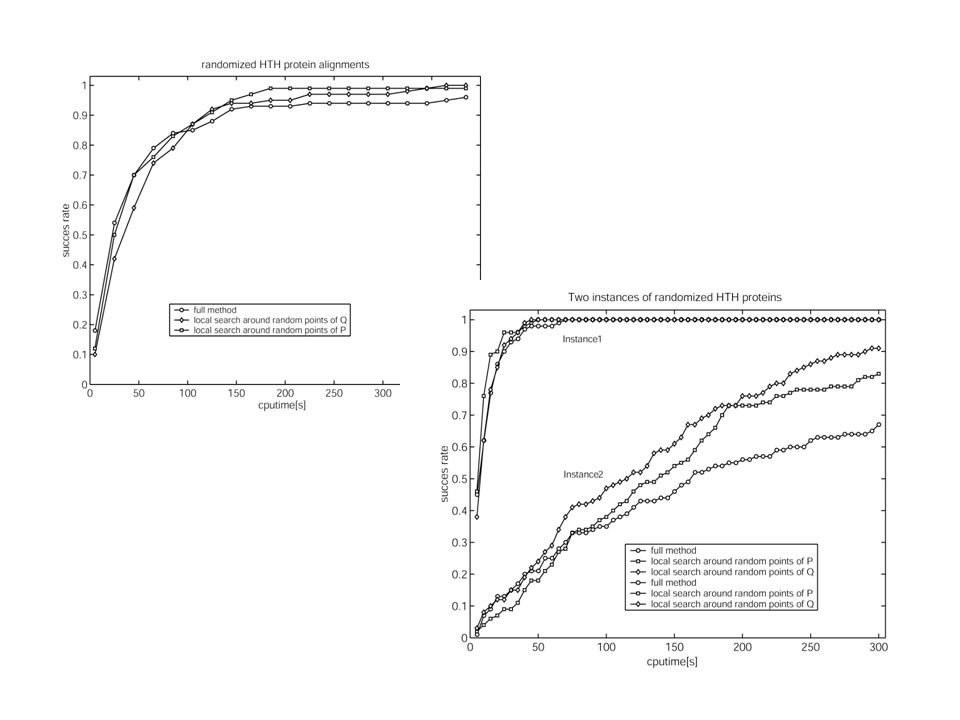
Performance de l'exploration: On mesure la capacité à trouver le meilleur point possible en un temps donné. Stratégies d'exploration: Recherche locale à partir de points aléatoires de P Gibbs sampler (hill-climbing stochastique), à partir de points aléatoires de P Recherche locale à partir de points aléatoires de Q Recherche locale à partir de points de Q choisis par un algorithme génétique (MoDEL).
, à partir de points aléatoires de P Recherche locale à partir de points aléatoires de Q Recherche locale à partir de points de Q choisis par un algorithme génétique (MoDEL)..")
11
Utilisation dun set de 16 protéines qui contiennent un domaine HTH. Ensemble de séquences construit à partir dinformation phylogénétique issue de: Rosinski & Atchley: 1999, molecular evolution of helix-turn- helix proteins Protéines distantes: faible conservation entre les sites, quasiment aucune conservation hors des sites.

12
Un 'succès' signifie que le maximum supposé a été atteint. le taux de succès est estimé sur 100 instances CPU: AMD athlon 1.5 GHz

14
Statistique sur le score Lentropie relative (ou log likelihood ratio) mesure une 'distance' entre les fréquences observées dans l'alignement, avec celles du background (=celles qu'on s'attend à trouver pour des alignements aléatoires). Cette mesure nous permet de comparer des alignements qui ont les mêmes paramètres libres, à savoir: le cardinal de lalphabet le background (paramètres dune distribution multinomiale) la longueur des sites le nombre de sites le nombre de libertés des sites (~longueur moyenne des séquences si on contraint le modèle à une occurrence par séquence)
 la longueur des sites le nombre de sites le nombre de libertés des sites (~longueur moyenne des séquences si on contraint le modèle à une occurrence par séquence).")
15
On ne peut donc pas comparer des alignements qui nont pas les mêmes paramètres (de longueurs différentes par ex.) Une solution est dutiliser une statistique comme la p-value ou le z-score, (le score du score en fonction des paramètres libres). Z-score: nombre décart-types à la moyenne. P-value: probabilité de lobservation quand lhypothèse nulle est vraie. Pval(x) = probabilité dobtenir un score supérieur ou égal à x par hasard. Hypothèse nulle: « Il ny a pas de motif » Randomisation des séquences (on garde les mêmes paramètres libres, mais on détruit les motifs potentiels).
 = probabilité dobtenir un score supérieur ou égal à x par hasard. Hypothèse nulle: « Il ny a pas de motif » Randomisation des séquences (on garde les mêmes paramètres libres, mais on détruit les motifs potentiels)..")
16
Statistique dalignement multiple local dans la littérature: Consensus: (Hertz & Stormo 1999) P-value: probabilité dobtenir un score supérieur ou égal, dans un ensemble aléatoire de même nombre de séquences et composition, mais de longueur infinie. MEME: (Bailey & Elkan 1995) E-value: nombre dalignements avec un score supérieur ou égal, que lon peut sattendre à trouver dans les séquences randomisées.
 E-value: nombre dalignements avec un score supérieur ou égal, que lon peut sattendre à trouver dans les séquences randomisées..")
17
HTHs: Résultat de MEME (serveur de linstitut pasteur)
")
18
HTHs: Résultats de Consensus: Capacité exploratoire limitée, ne trouve pas les sites. HTHs: Statistique de Consensus: (score optimisé par MoDEL) Estime une p-value de 1.84E-37 pour le score optimal: Estime une p-value de 1.7E-33 pour un score optimisé, avec des séquences randomisées. Avantage: rapide (peut être effectuée on the fly et permet de détecter la longueur optimale (en tout cas sur les HTHs) Désavantage: La valeur ne nous indique pas si on peut 'croire' à l'alignement produit.
 Estime une p-value de 1.84E-37 pour le score optimal: Estime une p-value de 1.7E-33 pour un score optimisé, avec des séquences randomisées. Avantage: rapide (peut être effectuée on the fly et permet de détecter la longueur optimale (en tout cas sur les HTHs) Désavantage: La valeur ne nous indique pas si on peut croire à l alignement produit..")
19
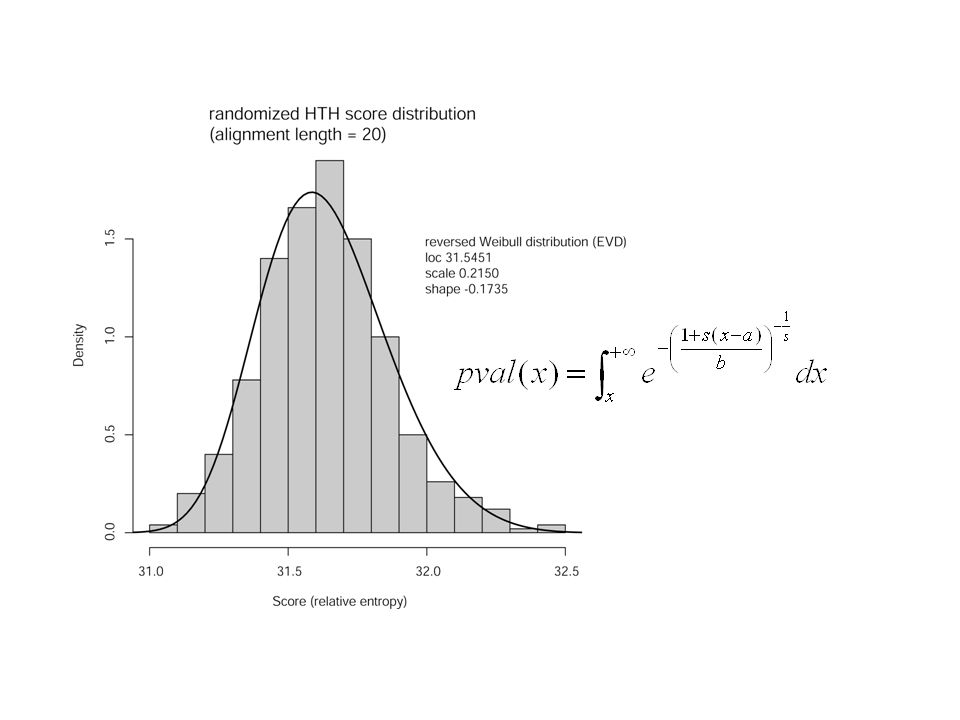
Si on veut une signification statistique, une méthode est destimer la probabilité dobserver un score optimisé sur les séquences randomisées, qui soit supérieur ou égal au score obtenu sur les séquences originales. On ne sait pas le faire analytiquement, donc: Génération déchantillons: On optimise un alignement sur un grand nombre de séquences randomisées (~200) Fitting de la distribution des scores avec une fonction de densité Calcul de la p-value à partir de la fonction de densité
 Fitting de la distribution des scores avec une fonction de densité Calcul de la p-value à partir de la fonction de densité.")
21
Comparaison d'alignements de différentes longueurs, par rapport à la p-value

22
Temps de calcul trop important pour une estimation on the fly ~1 à 3 heures de temps-cpu pour estimer la p-value dune longueur donnée Projet: Déterminer les valeurs des paramètres de l'EVD (loc, scale et shape), en fonction des paramètres libres (sans échantillonnage): Nombre de sites (nombre de séquences) Nombre de libertés des sites (longueur moyenne des séquences) Background (entropie des paramètres) Longueur de lalignement ~10 points par dimension: 10000 * 3 = 30000h temps-cpu pour échantillonner lespace.
, en fonction des paramètres libres (sans échantillonnage): Nombre de sites (nombre de séquences) Nombre de libertés des sites (longueur moyenne des séquences) Background (entropie des paramètres) Longueur de lalignement ~10 points par dimension: * 3 = 30000h temps-cpu pour échantillonner lespace.")
23
Comportement de loc, scale et shape en fonction de la longueur: