Télécharger la présentation
1
Isabelle Constant, Hôpital Armand Trousseau Paris, France
Paediatric haematology What the anaesthetist should know about … Coagulation problems Isabelle Constant, Hôpital Armand Trousseau Paris, France

2
. Plan Physiologie de l’hémostase (rappels)
Exploration de la coagulation Interrogatoire Examens biologiques Anomalies de l’hémostase primaire TP TCA Good morning Thank you, madam chairman and good afternoon ladies and gentleman. The intent of this talk is to present the basic principles of the haemostatic mechanism, then with this concepts in mind to develop an approach to the child who may have a bleeding history or an abnormal coagulation screening test. Finally, after reviewing some of the most commun bleeding disorders, I shall adress the issue of thrombotic disorders in children . Hypercoagulabilité Conclusion

3
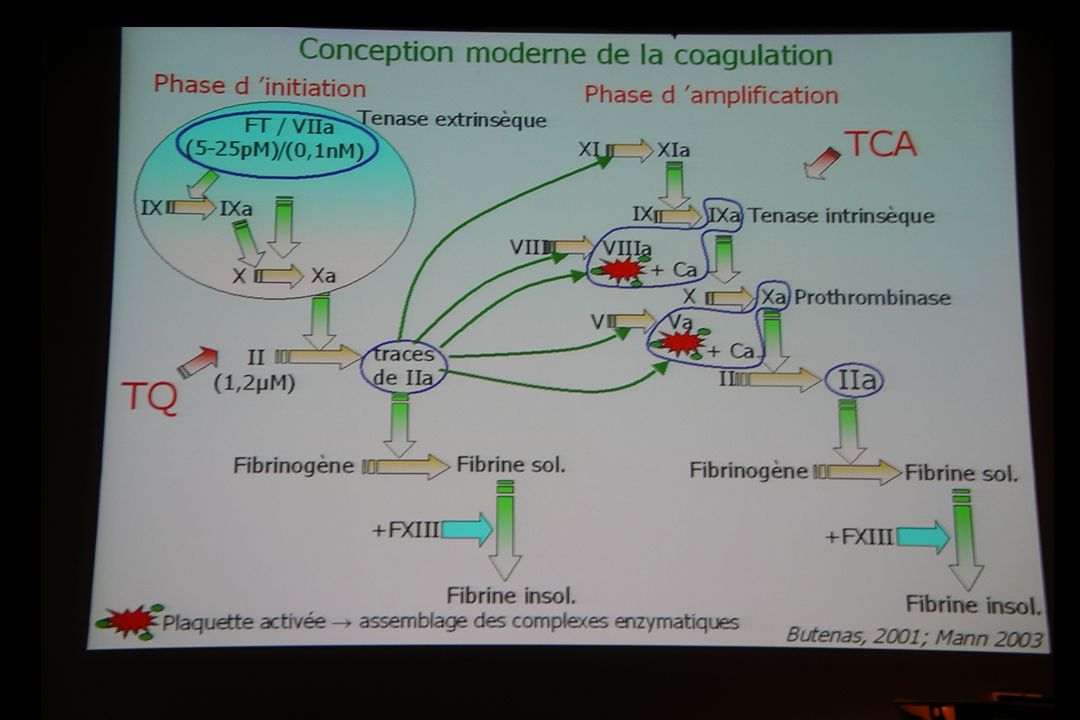
As you can see on this slide the hemostatic system is very simple.
Basically, The hemostatic mecanism includes three process, primary hemostasis, coagulation and fibrinolysis. What I would like you remember is that the activation of factor X can occur by two different reaction sequences : one from the tissue factor and the factor VII, and one from the complex factorVIII-factorIX. Once formed, Xa binds together with its cofactor factor Va, on the platelet phospholipid surface PF3 and together they activate factor II prothrombine to thrombine. And Thrombine converts fibrinogen to fibrin.

5
Spécificités de la coagulation chez le nouveau-né:
Hémostase primaire diminuée chez le nouveau-né Au delà de 10j, fonction et nombre de plaquettes normaux Activité des facteurs diminuée (sauf VIII et vW) Prolongation du TCA. Déficit des inhibiteurs naturels de la coagulation tels que l’ ATIII ou la protéine C Déficit de la fibrinolyse What about the specificities of the hemostatic system in neonate In Term infants and most preterm infants the platelet-vessel interaction is normal, as evidenced by normal bleeding time. Platelets number is also within the normal adult range. Many coagulation factors that mean II, VII, IX, X, XI, XII are decreased in activity. This global decrease leads to a prolongation of the most common coagulation screening tests such as PTT and PT. But the whole blood of preterm or term infants clots more quickly than the whole blood of children and adult ... This paradoxic hypercoagulability has been attributed to a deficiency of naturally occuring anticoagulants or protease inhibitors such as ATIII or protein C
 Prolongation du TCA. Déficit des inhibiteurs naturels de la coagulation tels que l’ ATIII ou la protéine C. Déficit de la fibrinolyse. What about the specificities of the hemostatic system in neonate. In Term infants and most preterm infants the platelet-vessel interaction is normal, as evidenced by normal bleeding time. Platelets number is also within the normal adult range. Many coagulation factors that mean II, VII, IX, X, XI, XII are decreased in activity. This global decrease leads to a prolongation of the most common coagulation screening tests such as PTT and PT. But the whole blood of preterm or term infants clots more quickly than the whole blood of children and adult ... This paradoxic hypercoagulability has been attributed to a deficiency of naturally occuring anticoagulants or protease inhibitors such as ATIII or protein C.")
6
Donc au total chez l ’enfant de moins de 6 mois
Le risque hémorragique spontané n ’est pas augmenté Mais le risque hémorragique provoqué en cas d ’anomalie constitutionnelle de l ’hémostase est majoré nécessité d ’un bilan d ’hémostase avant tout acte chirurgical même mineur Inhibiteurs Hypofibrinolyse Facteurs ETAT D ’EQUILIBRE INSTABLE

7
Toute hémorragie insolite bilan d ’hémostase
sec % 10 20 30 40 50 60 70 80 J2 J7 4A 3A 2A 1A 4M 8M FVIII TCA Marc J1 = Hémorragie digestive transfert en Réanimation (transfusions) Fibroscopie : lésion traumatique au niveau de l ’estomac Bilan d ’Hémostase = Plaquettes : /mm3 TCA : 48/35 sec TQ : 74 % Fibrinogène : 3,65 g/L F VIII : 50 % vWFAg : 92 % F IX : 50 % F XI : 42 % F XII : 39% Diagnostic : HEMOPHILIE A MINEURE Toute hémorragie insolite bilan d ’hémostase Tests globaux peu informatifs dosages spécifiques F VIII et FIX Interprétation des résultats en fonction des normes / l ’âge : taux augmenté du F VIII en période néonatale
 Fibroscopie : lésion traumatique au niveau. de l ’estomac. Bilan d ’Hémostase = Plaquettes : /mm3. TCA : 48/35 sec. TQ : 74 % Fibrinogène : 3,65 g/L. F VIII : 50 % vWFAg : 92 % F IX : 50 % F XI : 42 % F XII : 39% Diagnostic : HEMOPHILIE A MINEURE. Toute hémorragie insolite bilan d ’hémostase. Tests globaux peu informatifs. dosages spécifiques F VIII et FIX. Interprétation des résultats en fonction des normes / l ’âge : taux augmenté du F VIII en période néonatale.")
8
Consultation d’anesthésie Dépistage d’anomalies de l’hémostase
Examen clinique Interrogatoire Bilan préopératoire

9
Consultation d’anesthésie: interrogatoire
Antécédents familliaux de saignements Antécédents personnels de saignements Hémorragie importante après une chirurgie mineure Saignements lors d’une circoncision Hémarthrose Epistaxis Ecchymoses Pétéchies Saignement au cordon ombilical Prise médicamenteuse (Aspirine, AINS +++) Mais l’enfant peut ne pas avoir eu le temps de développer une histoire de saignements é Hémophilies Maladie de Von Willebrand Déficit en facteur XIII
 Mais l’enfant peut ne pas avoir eu le temps de développer une histoire de saignements. é. Hémophilies. Maladie de Von Willebrand. Déficit en facteur XIII.")
10
Bilan d’hémostase : recommandations
ANDEM 1992 : Bilan d’hémostase indiqué si examen clinique ou interrogatoire positifs ou enfant <âge de la marche GEHT 1993 : enfant + acte vulnérant bilan hémostase TCA, TQ, plaquettes ± TS ANAES 1998 : enfant > 3 ans, pas de bilan si interrogatoire fiable et chirurgie sans risque hémorragique

11
Questionnaire de dépistage d’anomalies de l’hémostase
L’enfant a-t-il saigné plus de 24 h voire nécessité une transfusion sanguine après un traumatisme ou un acte chirurgical mineur (circoncision, suture d’une plaie…)? L’enfant a-t-il saigné plus de 12h après la section du cordon ombilical (carnet de santé)? L’enfant a-t-il saigné dans la nuit voire au bout de 24h après une extraction dentaire? L’enfant a-t-il eu une ou plusieurs hématuries spontanées? Les questions 1 à 4 doivent être posées à la fratrie et aux parents. L’enfant présente-il fréquemment des ecchymoses sans cause apparente? L’enfant a-t-il déjà eu des épistaxis ayant nécessité un traitement chirurgical? Une extraction dentaire a-t-elle nécessité une suture chirurgicale? L’enfant a-t-il saigné plus de 15 min après une ponction veineuse? L’enfant a-t-il tendance au saignement continu? 1 réponse positive à une des questions 1 à 4, ou 2 réponses positives aux questions suivantes invite à prescrire une exploration de l’hémostase.
 L’enfant a-t-il saigné plus de 12h après la section du cordon ombilical (carnet de santé) L’enfant a-t-il saigné dans la nuit voire au bout de 24h après une extraction dentaire L’enfant a-t-il eu une ou plusieurs hématuries spontanées Les questions 1 à 4 doivent être posées à la fratrie et aux parents. L’enfant présente-il fréquemment des ecchymoses sans cause apparente L’enfant a-t-il déjà eu des épistaxis ayant nécessité un traitement chirurgical Une extraction dentaire a-t-elle nécessité une suture chirurgicale L’enfant a-t-il saigné plus de 15 min après une ponction veineuse L’enfant a-t-il tendance au saignement continu 1 réponse positive à une des questions 1 à 4, ou 2 réponses positives aux questions suivantes invite à prescrire une exploration de l’hémostase.")
12
Enquête hôpital Trousseau
Sur 1 an (2000), VG ou AVG Interrogatoire antécédents (ATCD) et signes cliniques (SC) en faveur d’une anomalie de l’hémostase Bilan d’hémostase systématique : TCA, TQ, plaquettes et TS (Ivy)(B1), et contrôlé si anomalies (B2) Un bilan ancien était accepté Anomalies hémostase notées et traitées si besoin Incidents hémorragiques notés
, VG ou AVG. Interrogatoire antécédents (ATCD) et signes cliniques (SC) en faveur d’une anomalie de l’hémostase. Bilan d’hémostase systématique : TCA, TQ, plaquettes et TS (Ivy)(B1), et contrôlé si anomalies (B2) Un bilan ancien était accepté. Anomalies hémostase notées et traitées si besoin. Incidents hémorragiques notés.")
13
Interro TS TCA Diagnostique Gr <3 ans + N Willebrand B+ O+ -
DDAVP vWFRCo=50% vWag=64% B+ vWag=44% O+ - vWag=65% Thrombopathie vWFRCo=96% vWag=63% >3 ans (n=500) vWFRCo=57% vWag=67% Maladie connue vWag=40% vWFRCo=125% vWag=90% A+ F XI = 5%
 vWFRCo=57% vWag=67% Maladie connue. vWag=40% vWFRCo=125% vWag=90% A+ F XI = 5%")
14
Hémorragies 16 patients (2%) ont présenté un saignement anormal per ou post opératoire Ces patients avaient tous des bilans preopératoires considérés comme normaux 8 patients (1%) ont été réopérés pour contrôle chirurgical de l’hémostase Aucun patient n’a été transfusé Parmi eux, on note : Une prise d’acide niflumique (2ans, saignement à J10) Une fonction hépatique limite : TQ:64%, VII+X:63% Hépatite B chronique active (8 ans saignements perop) Une thrombopathie de type pool vide partiel (7 ans, atcd chir sans pb, pas de bilan preop)
 ont été réopérés pour contrôle chirurgical de l’hémostase. Aucun patient n’a été transfusé. Parmi eux, on note : Une prise d’acide niflumique (2ans, saignement à J10) Une fonction hépatique limite : TQ:64%, VII+X:63% Hépatite B chronique active (8 ans saignements perop) Une thrombopathie de type pool vide partiel (7 ans, atcd chir sans pb, pas de bilan preop)")
15
Au total Un interrogatoire normal n’exclut pas une anomalie
Insuffisance des antécédents chirurgicaux 15% chez les <3 ans et 30% chez les >3 ans Fréquence du Willebrand 2 attitudes Minimaliste, bilan seulement si interrogatoire anormal (Ped Anaesth, 2002, 12:118) Maximaliste, bilan systématique <1 an, et après si chirurgie hémorragique ou ALR perimédullaire
 Maximaliste, bilan systématique <1 an, et après si chirurgie hémorragique ou ALR perimédullaire.")
16
Bilan biologique préopératoire
Enfant ayant déjà eu un contrôle de l’hémostase normal (au-delà de l’âge de 6 mois) pas de bilan Enfant ayant acquis l’âge de la marche et intervention mineure pas de bilan Dans les autres cas, bilan systématique NFS plaquettes TP, TCA Si interrogatoire positif exploration de l’hémostase primaire TS, PFA100
 pas de bilan. Enfant ayant acquis l’âge de la marche. et intervention mineure pas de bilan. Dans les autres cas, bilan systématique. NFS plaquettes. TP, TCA. Si interrogatoire positif exploration de l’hémostase primaire. TS, PFA100.")
17
Quid du temps de saignement ?
Le TS ne peut être coté que lorsque le médecin prescripteur justifie que l’interrogatoire du patient auquel il a procédé établi une tendance anormale au saignement (nomenclature des actes de biologie médicale) Non discriminant Peu fiable A abandonner en systématique Etude des fonctions plaquettaires (PFA) si doute sur l’hémostase primaire (interrogatoire, saignements non expliqués) Pathologies spécifiques Interactions pharmacologiques
 Non discriminant. Peu fiable. A abandonner en systématique. Etude des fonctions plaquettaires (PFA) si doute sur l’hémostase primaire (interrogatoire, saignements non expliqués) Pathologies spécifiques. Interactions pharmacologiques.")
18
La plupart des études montrent que environs 2% des enfants ont un bilan initial anormal …
… mais que chez la moitié de ces enfants le contrôle du bilan est normal. Burk et al 1992, Kang et al 1994 What about the results of the screening tests. Most of the studies show that approximately 2% of the children have initial laboratory abnormalities ... and half reverse to normal on repeated testing ! The initially large number of abnormal test may be due to laboratory or patient variation or transient viral illnesses. Then the first step in the investigation of abnormal coagulation screening tests should be repeated measurement, in approximately one week ’s time La première étape face à une hémostase anormale est le contrôle du bilan au mieux dans un délai de 1 semaine.

19
Taux minimum des facteurs de coagulation requis pour une hémostase chirurgicale
Fibrinogène g/l II, V, VIII, IX, X % XI % VII % XIII % XII, HMWK, Prekallicreine % Activité Willebrand % Plaquettes x 109/l Let’s look now at the minimal rate of the coagulation factors to allow normal hemostasis.. As you can see, only 30 to 40 % of activity is required for most of the factors to ensure a normal coagulation process. These values may be increased when the risk of bleeding of the surgical procedure is high. More over what you have to remember is that a deficiency in factor XII, in prekallicrein or in high-molecular-weight-kininogen has no consequences on the heamostatic process.

20
Bleeding time : Primary haemostasis
Lets go now to the bleeding time the template bleeding time is used as a screening test for primary hemostasis The primary hemostasis includes 3 processes which are illustrated on this slide. Vascular injury induces release of von Wlllebrand factor and exposes subendothelial collagen and provides a site for adhesion of platelets. Adhesion requires the interaction of platelet membrane glycoprotein Ib and the von Willebrand factor with the damaged endothelium. Aggregation of additionnal platelets is facilitated by agonists such as ADP, serotonin and thromboxan A2, through interaction of glycoprotein Ib-IIIa with fibrinogen.

21
Allongement du TS (Ivy method) ou PFA 100
Hémostase primaire Allongement du TS (Ivy method) ou PFA 100 Numération plaquettaire normale Maladie de Von Willebrand Anomalies fonctionnelles des plaquettes acquises (médicaments, IRC, atteintes virales…) Congenitales (Ehler-Danlos, Bernard Soulier, Glanzman’s disease…) Thrombopenie Purpura thrombocytopénique auto-immun (PTI) Destruction augmentée Production médullaire diminuée When blending is increased, you have to look at the platelet count, if the number of platelet is normal, you are before a functionnal platelet disorder and in this case the most frequent diagnosis is the von W disease. I’ll come on to this later in the following slides. The thrombopathy induced by drug absorption is usually identify by asking parents. The congenital thrombopathy are very rare. Now if there is a thrombopenia, the first diagnosis is The autoimmune thrombocytopenic purpura (ITP).this the most frequent desorder of primary hemostasis in children with no personal or familial history of bleeding. ITP in children usually follows viral infection with the production of virus induced antibodies that recognize glycoproteins on the surface of the platelet And cause rapid removal of the platelet from the circulation. This disease is generally already diagnosed by the paediatician. The other causes of thrombopenia are much less frequent. You may challenge me and asking if the bleeding time is really necessairy. I know it is not routinely performed and its usefulness had been challenged at least in adults. Nevertheless in our institution, we carry on to perform this test to study the primary hemostasis Clinical usefulness of bleeding time is challenged (Editorial Lancet 1991, O’Kelly et al BJA 1992)
 ou PFA 100. Numération plaquettaire normale. Maladie de Von Willebrand. Anomalies fonctionnelles des plaquettes. acquises (médicaments, IRC, atteintes virales…) Congenitales (Ehler-Danlos, Bernard Soulier, Glanzman’s disease…) Thrombopenie. Purpura thrombocytopénique auto-immun (PTI) Destruction augmentée. Production médullaire diminuée. When blending is increased, you have to look at the platelet count, if the number of platelet is normal, you are before a functionnal platelet disorder and in this case the most frequent diagnosis is the von W disease. I’ll come on to this later in the following slides. The thrombopathy induced by drug absorption is usually identify by asking parents. The congenital thrombopathy are very rare. Now if there is a thrombopenia, the first diagnosis is The autoimmune thrombocytopenic purpura (ITP).this the most frequent desorder of primary hemostasis in children with no personal or familial history of bleeding. ITP in children usually follows viral infection with the production of virus induced antibodies that recognize glycoproteins on the surface of the platelet And cause rapid removal of the platelet from the circulation. This disease is generally already diagnosed by the paediatician. The other causes of thrombopenia are much less frequent. You may challenge me and asking if the bleeding time is really necessairy. I know it is not routinely performed and its usefulness had been challenged at least in adults. Nevertheless in our institution, we carry on to perform this test to study the primary hemostasis. Clinical usefulness of bleeding time is challenged. (Editorial Lancet 1991, O’Kelly et al BJA 1992)")
22
Interactions pharmacologiques
Aspirine AINS (+++) Seul le délai de 8 à 10 jours est garant de la restauration de l’hémostase primaire
 Seul le délai de 8 à 10 jours est garant de la restauration de l’hémostase primaire.")
23
Le cauchemar du clinicien
Maladie de Willebrand = Le cauchemar du clinicien Incidence élevée Risque de saignements per et postopératoires Problème du Willebrand modéré

24
Facteur von Willebrand : 2 roles majeurs
Let’s go now to the von Willebrand disease. Von Willebrand factor serves two key roles in normal hemostasis. A simplified schema of these roles is shown on this slide. First point:Circulating VWF does not bind platelets,but exposure to the subendothelial matrix induces a structural change in vWF, allowing it to bind to its receptor platelet glycoprotein Ib. The interaction of vWF and its platelet receptor leads to platelet adhesion and activation. Second point: vWF is the carrier protein for FVIII in the plasma. VWF protects FVIII from proteolytic degradation in the plasma, especially by activated protein C. In the absence of VWF, FVIII has a markedly reduced half life. This may explain the deficiency of FVIII observed in vW disease, and consequently the frequently associated prolongation of the PPT. When the vWf activity is restored the deficiency of fact VIII disappears and the PPT is corrected

25
Maladie de Von Willebrand
1% de la population polymorphisme genetique Type Déficit 1 Quantitatif (partiel) Le plus commun 2A Anomalie qualitative du vWF 2B Anomalie qualitative du vWF 2N Anomalie de la liaison au facteur VIII phénotype hémophilie 2M Anomalie fonctionnelle du vWF 3 Quantitatif (majeur) vW disease is the most common congenital bleeding disorder, affecting at least 1% of the population. Several defects of the vWF gene are described leading to a complex genetic polymorphism. The new classification that you can see on the slide, is based on the type of the molecular abnormalities. Basically, type 1 which is the most common form of the disorder is characterised by a mild to moderate decrease in plasma levels of vWF, with a proportionate decrease of the fact VIII All Type 2 are characterised by partial defect of vWF, leading to abnormal vWF function.( In type 2N, the defect is located at the level of FVIII binding region, leading to a rapid proteolysis of the FVIII. Affected patients have a mild hemophilia phenotype, but unlike patients with hemophilia, their disorder autosomal inheritance) type 3 is a severe bleeding disorder with major deficit of both primary and secondary hemostasis. Plasma levels of fVIII are very low, whereas those of vWF are virtually undetectable. Sadler, 1994
 Le plus commun. 2A Anomalie qualitative du vWF. 2B Anomalie qualitative du vWF. 2N Anomalie de la liaison au facteur VIII phénotype hémophilie. 2M Anomalie fonctionnelle du vWF. 3 Quantitatif (majeur) vW disease is the most common congenital bleeding disorder, affecting at least 1% of the population. Several defects of the vWF gene are described leading to a complex genetic polymorphism. The new classification that you can see on the slide, is based on the type of the molecular abnormalities. Basically, type 1 which is the most common form of the disorder is characterised by a mild to moderate decrease in plasma levels of vWF, with a proportionate decrease of the fact VIII. All Type 2 are characterised by partial defect of vWF, leading to abnormal vWF function.( In type 2N, the defect is located at the level of FVIII binding region, leading to a rapid proteolysis of the FVIII. Affected patients have a mild hemophilia phenotype, but unlike patients with hemophilia, their disorder autosomal inheritance) type 3 is a severe bleeding disorder with major deficit of both primary and secondary hemostasis. Plasma levels of fVIII are very low, whereas those of vWF are virtually undetectable. Sadler,")
26
Desmopressine (DDAVP) et maladie de Willebrand
DDAVP multiplie par 2 ou 3 les concentrations de vWF et FVIII chez les patients répondeurs Indiquée dans les Willebrand type I Utile pour préparer un patient répondeur avant la chirurgie contre-indiquée chez l’enfant de moins de 2 ans. L’enfant doit être testé avant le jour de la chirurgie (dose test 0.2 mcg/kg en 30 min) Si le patient est répondeur, perfusion de DDAVP ( mcg/kg), 1 heure avant la chirurgie, éventuellement répétée 12 h après Hyponatrémie = iatrogènicité Desmopressine is a synthetic analog of antidiuretic hormone modified to have less pressor activity and a longer half life than the original compound. At appropriate dose desmopressin causes a twofold to threefold increase in plasma vWF and FVIII concentration in responsive patients. A principal mecanism of desmopressin action is relaese of vWF from endothelial storage sites. Desmopressin is indicated in typeI and in some type 2A. Desmopressin is not effective in the other type. Desmopressin can be usefull to prepare a patient of more then 2 years of age for surgery. To know if the child is responsive to desmopressin, The child should be tested prior to surgery If the patient is responsible, desmopressin is given 1 hour before the surgery, and repeated every 12 h with a maximum of four injections Hyponatremia is the main side effect.
 Si le patient est répondeur, perfusion de DDAVP ( mcg/kg), 1 heure avant la chirurgie, éventuellement répétée 12 h après. Hyponatrémie = iatrogènicité. Desmopressine is a synthetic analog of antidiuretic hormone modified to have less pressor activity and a longer half life than the original compound. At appropriate dose desmopressin causes a twofold to threefold increase in plasma vWF and FVIII concentration in responsive patients. A principal mecanism of desmopressin action is relaese of vWF from endothelial storage sites. Desmopressin is indicated in typeI and in some type 2A. Desmopressin is not effective in the other type. Desmopressin can be usefull to prepare a patient of more then 2 years of age for surgery. To know if the child is responsive to desmopressin, The child should be tested prior to surgery. If the patient is responsible, desmopressin is given 1 hour before the surgery, and repeated every 12 h with a maximum of four injections. Hyponatremia is the main side effect.")
27
Let me now turn to the prothrombone time PT or Quick test
On this slide, you can see in blue the area investigated by the PT. This test measures thrombin generation in the extrinsic pathway and is a function of fact II, V, X and VII.

28
Allongement du TQ (1) Déficit en vitamine K Fréquent
Antibiothérapies itératives, exclusion digestive Supplémentation par voie orale quelques jours avant la chirurgie Limites ?

29
Allongement du TQ (2) Déficit en facteur VII
Non exceptionnel (1/ ) >15%, pas de supplémentation initiale, wait and see
 >15%, pas de supplémentation initiale, wait and see.")
30
TCA Let’s go now to the partial prothrombine time or PTT
which measures thrombin generation in the intrinsic pathway and is a function of all the coagulation factors except factor VII

31
Déficit en facteur XII (Hageman trait) Maladie de von Willebrand
Allongement du TCA Anticoagulants circulants (antiphospholipides) Déficit en facteur XII (Hageman trait) Maladie de von Willebrand Déficit congénital en facteur VIII (Hémophilie classique) Déficit congénital en facteur IX (Christmas disease) Déficit congénital en facteur XI (rare) Déficit en prekallikreine plasmatique et déficit en kininogène de haut poids moléculaire. What about a prolonged PPT. In order of frequency you could find Firstly : Circulating anticoagulants which occur in children mostly after viral infection. These anticoagulants are directed against phospholipids and may be detected by prolongation of the PPT which does not correct with mixing with normal plasma. Usually this circulating anticoagulants are not associated with significant haemorrhage. And the hematologist give you the green light for the surgery. The Second cause is the Deficiency in factor 12, and again that is not an haemorrhagic disorder, but it is mentioned here to call attention to marked and frequent laboratory abnormalities associated with this hereditary trait.( Since these patients are not subject to abnormal bleeding, specific identification of their deficiency is important because this is usually found as an abnormal result in a screening test. ) The vWD is the third hypothesis in frequency, but this is the first real hemorragic disorder before a prolonged¨PPT. As I have previously said, this prolongation is due to a functionnal defect of the factor VIII let’s now look at the question of hemophilias for which the PTT is the most sensitive screening test. The PTT is elevated when level of factor VIII activity is below 40% of normal activity. diagnosis is confirmed by a specific assay for factor VIII activity.or for factor IX A Deficiency of factor XI could also be detected but it’s very rare. So, basically even if a prolonged PPT is frequently not associated with an increase of the risk of bleeding, that may lead to the diagnosis of vWF or hemophilia
 Déficit en facteur XII (Hageman trait) Maladie de von Willebrand. Déficit congénital en facteur VIII (Hémophilie classique) Déficit congénital en facteur IX (Christmas disease) Déficit congénital en facteur XI (rare) Déficit en prekallikreine plasmatique et déficit en kininogène de haut poids moléculaire. What about a prolonged PPT. In order of frequency you could find. Firstly : Circulating anticoagulants which occur in children mostly after viral infection. These anticoagulants are directed against phospholipids and may be detected by prolongation of the PPT which does not correct with mixing with normal plasma. Usually this circulating anticoagulants are not associated with significant haemorrhage. And the hematologist give you the green light for the surgery. The Second cause is the Deficiency in factor 12, and again that is not an haemorrhagic disorder, but it is mentioned here to call attention to marked and frequent laboratory abnormalities associated with this hereditary trait.( Since these patients are not subject to abnormal bleeding, specific identification of their deficiency is important because this is usually found as an abnormal result in a screening test. ) The vWD is the third hypothesis in frequency, but this is the first real hemorragic disorder before a prolonged¨PPT. As I have previously said, this prolongation is due to a functionnal defect of the factor VIII. let’s now look at the question of hemophilias for which the PTT is the most sensitive screening test. The PTT is elevated when level of factor VIII activity is below 40% of normal activity. diagnosis is confirmed by a specific assay for factor VIII activity.or for factor IX. A Deficiency of factor XI could also be detected but it’s very rare. So, basically even if a prolonged PPT is frequently not associated with an increase of the risk of bleeding, that may lead to the diagnosis of vWF or hemophilia.")
32
Allongement du TCA (3) Déficit en FVIII FVIII Sévérité Traitement
80 % de tous les patients porteurs d’une hémophilie Incidence estimée de 1/10 000 Taux d’ antigène vW normal La sévérité des manifestations cliniques est corrélée au taux de facteur VIII (# constant pour un patient donné) FVIII Sévérité Traitement < 1% Severe FVIII concentrés 1-6 % Modérée FVIII concentrés 6- 30 % Mineure FVIII concentrés or DDAVP ( mcg/kg en 30 min)
 FVIII Sévérité Traitement. < 1% Severe FVIII concentrés. 1-6 % Modérée FVIII concentrés % Mineure FVIII concentrés. or DDAVP. ( mcg/kg en 30 min)")
33
Incidence très faible des thromboses chez l’enfant : 0.07 /100 000
Facteurs acquis Stase vasculaire (cathéter veineux central, immobilisation…) Maladie inflammatoire sévère Anticorps antiphospholipides Déficits héréditaires des inhibiteurs de la coagulation ATIII (<85%) Proteine C (<65%) Proteine S (<65%) Dysfibrinogenemie facteur V leiden, facteur II Leiden Moving on now to the last question of the thrombosis in children In fact thrombosis is fortunately very rare in children Some factors are known to increase the risk of thrombosis such as vascular stasis, severe inflamatory disease or sickle cell disease but In most cases the thrombosis occurs when there is the conjunction of one of these previous factors and an hereditary deficiency of natural anticoagulants such as mainly ATIII, Prot C or Prot S.
 Maladie inflammatoire sévère. Anticorps antiphospholipides. Déficits héréditaires des inhibiteurs de la coagulation. ATIII (<85%) Proteine C (<65%) Proteine S (<65%) Dysfibrinogenemie. facteur V leiden, facteur II Leiden. Moving on now to the last question of the thrombosis in children. In fact thrombosis is fortunately very rare in children. Some factors are known to increase the risk of thrombosis such as vascular stasis, severe inflamatory disease or sickle cell disease but In most cases the thrombosis occurs when there is the conjunction of one of these previous factors and an hereditary deficiency of natural anticoagulants such as mainly ATIII, Prot C or Prot S.")
34
Déficit en facteur XIII
Deux anomalies rares, héréditaires et congénitales, de l’hémostase dans lesquelles les examens de l’hémostase sont normaux Déficit en Alpha2-antiplasmin Déficit en facteur XIII This slide just to tell you that there are two rare congenital hereditary hemorragic disorders, in which the hemostatic screening tests are normal. Then when clinical history is positive despite the usual test of hemostatis are normal you and your hamatologist should think about the Quand l’histoire clinique décrit un sydrôme hémorragique et que les tests d’hémostases reviennent normaux…

35
To conclude this talk I would like to say that the management of an abnormal coagulation screening test or the management of an abnormal bleeding should be based on a consensus between the anesthetist the heamatologist and the surgeon.

36
Conduite à tenir devant une thrombopénie
Vérifier la réalité de la thrombopénie Morphologie et volume plaquettaires Existe-t-il un contexte clinique évocateur ? Prise médicamenteuse Infection Hypersplénisme ou consommation

37
Enfant de 4 ans Asthénie, fièvre adénopathies GB: 41 G/l Hb: 10 g/dl VGM 80 fl Plq: 80 G/l 85 % lymphocytes atypiques Syndrome mononucléosique sérologies virales Diagnostic d’une thrombopénie virale Auto anticorps transitoires Infection ou vaccination par: rubéole, varicelle, rougeole oreillons, MNI, CMV, hépatite B ou C, VIH…

38
Enfant pâle, présentant un purpura diffus.
Petit garçon de 3 ans admis aux urgences pour diarrhée sanglante, douleurs abdominales, oligurie et fièvre à 38° depuis 3 jours. Enfant pâle, présentant un purpura diffus. GB: 12 G/L Hb: 6,0 g/dL VGM 80 fl PLq: 31 G/L Rétic: 120 G/l Hapto < 0.1 g/l Schizocytes: 5% Insuffisance rénale Anémie hémolytique (schizocytes) régénérative avec thrombopénie, IRA fonctionnelle Diagnostic d ’un syndrome hémolytique et urémique (SHU)
 régénérative avec. thrombopénie, IRA fonctionnelle. Diagnostic d ’un syndrome hémolytique et urémique (SHU)")
39
Conduite à tenir devant une thrombopénie
Vérifier la réalité de la thrombopénie Morphologie et volume plaquettaires Existe-t-il un contexte clinique évocateur ? Prise médicamenteuse Infection Hypersplénisme ou consommation Absence de contexte évocateur Myélogramme Cause périphérique: diagnostic d ’un PTI Cause centrale

40
Enfant de 3 ans Consulte pour purpura pétéchial, diffus, d ’apparition récente Pas d ’ATCD hémorragiques GB: 8 G/l Hb: 11 g/dl Plq: < 10 G/l Absence d’organomégalie Pas de fièvre Frottis: RAS Absence de contexte clinique évocateur Myélogramme

41
Moelle normale, très nombreux mégacaryocytes
Thrombopénie périphérique Diagnostic d’un PTI: purpura thrombopénique auto immun Destruction des plaquettes par macrophages Pic de fréquence entre 2 et 5 ans Risque d ’hémorragie intra-crânienne 1%

42
Myélogramme anormal: Infiltration médullaire: LA, LMMC, lymphomes, cancers Aplasie débutante: Fanconi, aplasie idiopathique SAM: syndrome d’activation macrophagique Amégacaryocytose immunologique Maladie métabolique: acidémies-aciduries organiques acidémie propionique, acidémie méthyl malonique thrombopénie souvent élément révélateur de la maladie

43
Thrombopénies et risque hémorragique
Recommandations (College of American Pathologists,1994) Si procédure invasive <5 G Extrêmement vraisemblable Entre 5 et 10 G/l Très vraisemblable Entre 10 et 50 G/l Variable >50 G Peu vraisemblable En pratique Chirurgie Taux de plaquettes à remonter au dessus de si neurochirurgie, chirurgie ORL et ophtalmique Anesthésies rachidienne
 Si procédure invasive. <5 G Extrêmement vraisemblable. Entre 5 et 10 G/l Très vraisemblable. Entre 10 et 50 G/l Variable. >50 G Peu vraisemblable. En pratique. Chirurgie Taux de plaquettes à remonter au dessus de si neurochirurgie, chirurgie ORL et ophtalmique. Anesthésies rachidienne")
44
Thrombopénies Mélange de concentrés de plaquettes standard déleucocytés (CPS) 1 CPS ( x1011) pour 7 kg de poids Concentrés de plaquettes d’aphérèse déleucocytés (CPA) 1 CPA # 4 CPS DDAVP Immunoglobulines
 1 CPA # 4 CPS. DDAVP. Immunoglobulines.")
45
Conclusions Difficultés spécifiques à l ’étude de l ’hémostase pédiatrique: Qualité des prélèvements Limites des tests de dépistage actuels Interprétation des résultats en fonction de l ’âge et du terme La qualité du dialogue biologiste / clinicien est capitale pour orienter nos investigations Notre rôle de biologiste, dans le domaine de l ’hémostase tout particulièrement, est de participer au diagnostic et d ’apporter une aide à la thérapeutique

46
TAUX D’ HEMOGLOBINE POUR UNE DELIVRANCE D’OXYGENE EQUIVALENTE
Hb Normale Adulte Nourrisson 5, ,5 7, ,2 10 (>3 mois) Nouveau-né 10, ,7 13, ,7 17 (<2 mois)
 Nouveau-né 10,3 11,7 13,2 14,7 17. (<2 mois)")
















>")
>")


