Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Les données et les banques de données

2
Les données chromosome

3
Les données position gène chromosome

4
Les données position gène structure séquence chromosome Intron Exon
Motifs régulateurs Codon start Codon stop

5
Les données position gène structure séquence Quantification séquence
chromosome structure séquence Intron Exon Motifs régulateurs Codon start Codon stop ARN pré-messager Quantification expression ADNc ESTs ARN messager séquence AAAAA

6
Les données position gène structure séquence Quantification expression
chromosome structure séquence Intron Exon Codon start Codon stop Motifs régulateurs ARN pré-messager Quantification expression ADNc ESTs ARN messager séquence AAAAA Protéine séquence structure fonctions

7
Les banques de données La principale mission des banques est de rendre publiques les données issues du séquençage Il existe deux types de banques de données : banques généralistes : collecte de données la plus exhaustive possible banques spécialisées : établies autour d’une thématique principale

8
Les banques de données généralistes
Les banques de séquences nucléiques les séquences nucléiques peuvent être de plusieurs natures : ADN génomique, ADNc ... EMBL pour European Moleculary Biology Library créée et financée en 1980 parl ’EMBO diffusée actuellement par l ’EBI (Angleterre) GENBANK créée en 1982 par la société IntelliGenetics et diffusée actuellement par le NCBI (USA) DDBJ pour DNA Data Bank of Japan Créée en 1986 par le NIG Japon Echanges systématiques des données depuis 1987 Mise en place d’un système de conventions communes
 GENBANK. créée en 1982 par la société IntelliGenetics et. diffusée actuellement par le NCBI (USA) DDBJ pour DNA Data Bank of Japan. Créée en 1986 par le NIG Japon. Echanges systématiques des données depuis Mise en place d’un système de conventions communes.")
13
Les banques de données généralistes (2)
Les banques de séquences protéiques une protéine peut être obtenue de deux manières différentes : 1. in silico : déduite de la séquence nucléique par simple traduction 2. isolée à partir de la cellule et séquencée PIR-NBRF créée en 1984 par la NBRF SWISSPROT créée en 1986 à l ’université de Genève maintenue depuis 1987 entre cette université et l ’EBI

15
Les défauts des banques de données
Principal défaut : le manque de vérification des données soumises 2. Hétérogénéité dans la nature de séquences on trouve : ADN nucléaire, ADN mitochondrial, ADN chloroplastique, ARN messager, ARN de transfert, chromosomes entiers… 3. Variabilité de l’état des connaissances sur les séquences travail expérimental ou prédiction bioinformatique 4. Erreurs dans les séquences 5. Biais d’échantillonnage toutes les espèces ne sont pas représentées tous les gènes d ’une espèces ne sont pas présents il existe une redondance des données

16
Les banques spécialisées
Devant la croissance exponentielle et l’hétérogénéité des séquences des banques spécialisées se sont constituées autour de thématiques biologiques particulières. Exemple : les motifs spécifiques d ’une séquence nucléique ou protéique sites ayant une activité biologique ( TATAbox, site de fixation de l ’ATP) Les bases de motifs nucléiques (éléments régulateurs …) TRANSFAC (1993), TFD (1994) Les bases de motifs protéiques PROSITE (1993), BLOCK (1991) Toutes ces données représentent un espace de connaissances de références à partir desquelles on tentera d ’identifier les séquences des gènes inconnus, issues du séquençage.
 Les bases de motifs nucléiques (éléments régulateurs …) TRANSFAC (1993), TFD (1994) Les bases de motifs protéiques. PROSITE (1993), BLOCK (1991) Toutes ces données représentent un espace de connaissances de. références à partir desquelles on tentera d ’identifier les séquences. des gènes inconnus, issues du séquençage.")
21
Manipulation des données
Les séquences sont stockées en général sous forme de fichiers texte qui peuvent être soit des fichiers personnels, soit des fichiers publics accessibles par des programmes interfaces (SRS, GCG, Acnuc). Le format correspond à l'ensemble des règles (contraintes) de présentation auxquelles sont soumises la ou les séquences dans un fichier donné. Ainsi, le format permet donc : > une mise en forme automatisée, > le stockage homogène de l'information, > le traitement informatique ultérieur de l'information. Pour lire et traiter les séquences, les logiciels d'analyse autorisent un ou plusieurs formats des données. Partie I du TD
. Le format correspond à l ensemble des règles (contraintes) de présentation. auxquelles sont soumises la ou les séquences dans un fichier donné. Ainsi, le format permet donc : > une mise en forme automatisée, > le stockage homogène de l information, > le traitement informatique ultérieur de l information. Pour lire et traiter les séquences, les logiciels d analyse. autorisent un ou plusieurs formats des données. Partie I du TD.")
22
Annotations des séquences

23
L’annotation Séquences informations biologiques Annotation structurale
localisation des gènes structure des gènes régions codantes positions des motifs régulateurs Expérimentations humides Annotation fonctionnelle fonction biochimique fonctions biologiques régulation et intéractions expression Prédictions in silico

24
Détection de gènes eucaryotes ab initio
La recherche de gène dans le génome d'espèce eucaryote est complexe : 1. les régions codantes sont morcelées par la présence d'introns 2. les régions codantes représentent moins de 5% du génome eucaryote. La prédiction des gènes de génome eucaryote peut être appréhendée selon trois approches: 1. basée sur la similarité de séquence 2. ab initio 3. génomique comparative

25
Similarité de séquence
Pour la recherche des gènes, elle peut être utilisée selon trois voies : - comparaison directe de la séquence génomique avec des ESTs détermination des séquences codantes et des limites intron/exon - comparaison des six cadres de lecture de la séquence contre des protéines. - comparaison entre les différentes traductions de la séquence génomique et celles de banque de données génomiques et d’ADNc. similarité de séquence : méthode puissante mais non infaillible

26
Comparaison de séquences
Séquence brute : à quoi elle correspond ? actcttctggtccccacagactcagagagaacccaccatggtgctgtctcctgccgacaagaccaacgtcaaggccgcctggggtaaggt cggcgcgcacgctggcgagtatggtgcggaggccctggagaggatgttcctgtccttccccaccaccaagacctacttcccgcacttcga cctgagccacggctctgcccaggttaagggccacggcaagaaggtggccgacgcgctgaccaacgccgtggcgcacgtggacgacatgcc caacgcgctgtccgccctgagcgacctgcacgcgcacaagcttcgggtggacccggtcaacttcaagctcctaagccactgcctgctggt gaccctggccgcccacctccccgccgagttcacccctgcggtgcacgcctccctggacaagttcctggcttctgtgagcaccgtgctgac ctccaaataccgttaagctggagcctcggtggccatgcttcttgccccttgggcctccccccagcccctcctccccttcctgcacccgta cccccgtggtctttgaataaagtctgagtgggcggc 1. Ressemblance avec d’autres séquences déjà connues? 2. Trouver toutes les séquences d’une même famille 3. Rechercher toutes les séquences qui contiennent un motif donné

27
Dot Plot Les dot plots sont utilisés :
- pour comparer visuellement deux séquences et détecter les régions ayant une forte similarité Les deux séquences sont placées le long des axes d ’un graphique. 2. L ’intersection de chaque ligne et colonne est marquée d ’un point si la lettre est identique dans les deux séquences Une suite de points sur la diagonale indique : les régions de similarité entre les deux séquences.

28
Dot Plot Avec des séquences réelles, les motifs ne sont pas si évidents !

29
Alignement de séquences
Objectif : essayer de faire le maximum d ’appariements entre deux séquences - avoir le plus d’identité entre les séquences X et Y Lorsque deux séquences sont comparées: on observe des substitutions des insertions et des délétions Exemple 1: S1 : TCAGACGATTG n=11 S2 : TCGGAGCTG m = 9 alignement 1 identité x substitution y Indel z TCAG-ACG-ATTG TC-GGA-GC-T-G alignement 2 TCAGACGATTG TCGGAGCTG alignement 3 TCAG ACGATTG TC GGA GCTG

30
Quel est le meilleur alignement ?
Ce qu’on veut, c’est le maximum de bases identiques et le minimum de substitutions et indels donc 1. On va pénaliser les substitutions et les insertions/délétions (w) 2. On va rechercher un coût minimal pour l’alignement coût = w substitution * y + w indel * z pour les alignements précédents : si w substitution = 1 et w indel = 2 alignement 1 Coût = 1 *0 + 6*2 = 12 alignement 2 Coût = 1*5 + 2*2 = 9 <- alignement qui a le moins de coût : c’ est le meilleur alignement 3 Coût = 1*2 + 2*4 = 10
 2. On va rechercher un coût minimal pour l’alignement. coût = w substitution * y + w indel * z. pour les alignements précédents : si w substitution = 1 et w indel = 2. alignement 1. Coût = 1 *0 + 6*2 = 12. alignement 2. Coût = 1*5 + 2*2 = 9 <- alignement qui a le moins de coût : c’ est le meilleur. alignement 3. Coût = 1*2 + 2*4 = 10.")
31
Recherche de similarité globale ou locale
Les finalités de ces deux types de recherche sont très différentes. L'alignement global (Needelman & Wunch) : comparer des séquences homologues (apparentées) sur toute leur longueur. L'alignement local (Smith & Waterman, BLAST, FASTA) est conçu rechercher dans la séquence A des régions semblables à la séquence B
 : comparer des séquences homologues (apparentées) sur toute leur longueur. L alignement local (Smith & Waterman, BLAST, FASTA) est conçu. rechercher dans la séquence A des régions semblables à la séquence B.")
32
Les matrices de substitution
Pour aligner deux séquences, il faut évaluer leur similarité en attribuant un score grâce à des matrices de scores. On distingue deux types de scores: - le score élémentaire qui est la valeur donnée directement dans la matrice - le score global qui est calculé comme la somme des scores élémentaires Exemple: A T C G A T C G Score élémentaire ATGGCTAGAACT TACGGCTTAGCTA Score global = 5

33
Exemples de matrices protéiques
1. Les matrices PAM (Point Accepted Mutation) M. Dayhoff années 70 Obtenues par l’étude de 71 familles de protéines (1300 séquences) Elles donnent les scores de similarité obtenus en fonction du nombre de mutation pour une séquence de longueur 100 aa. Ex : PAM1 : 1 mutation entre deux séquences de 100 aa (~= 100 % identité) les deux séquences sont pratiquement identiques PAM250 : 250 mutations (~= 20% identité) 2. Les matrices de type BLOSUM (BLOcks SUbstitution Matrix) obtenues à partir de motifs conservés entre des familles de protéines les matrices BLOSUM plus récentes donnent en général de meilleurs résultats 3. Les matrices liées aux caractéristiques physico-chimique ex: caractère hydrophile ou hydrophobe des protéines BLOSUM 80 BLOSUM BLOSUM45 PAM 1 PAM PAM 250 Peu différent très différent
 M. Dayhoff années 70. Obtenues par l’étude de 71 familles de protéines (1300 séquences) Elles donnent les scores de similarité obtenus en fonction du nombre de mutation. pour une séquence de longueur 100 aa. Ex : PAM1 : 1 mutation entre deux séquences de 100 aa (~= 100 % identité) les deux séquences sont pratiquement identiques. PAM250 : 250 mutations (~= 20% identité) 2. Les matrices de type BLOSUM (BLOcks SUbstitution Matrix) obtenues à partir de motifs conservés entre des familles de protéines. les matrices BLOSUM plus récentes donnent en général de meilleurs résultats. 3. Les matrices liées aux caractéristiques physico-chimique. ex: caractère hydrophile ou hydrophobe des protéines. BLOSUM 80 BLOSUM 62 BLOSUM45. PAM 1 PAM 120 PAM 250. Peu différent. très différent.")
34
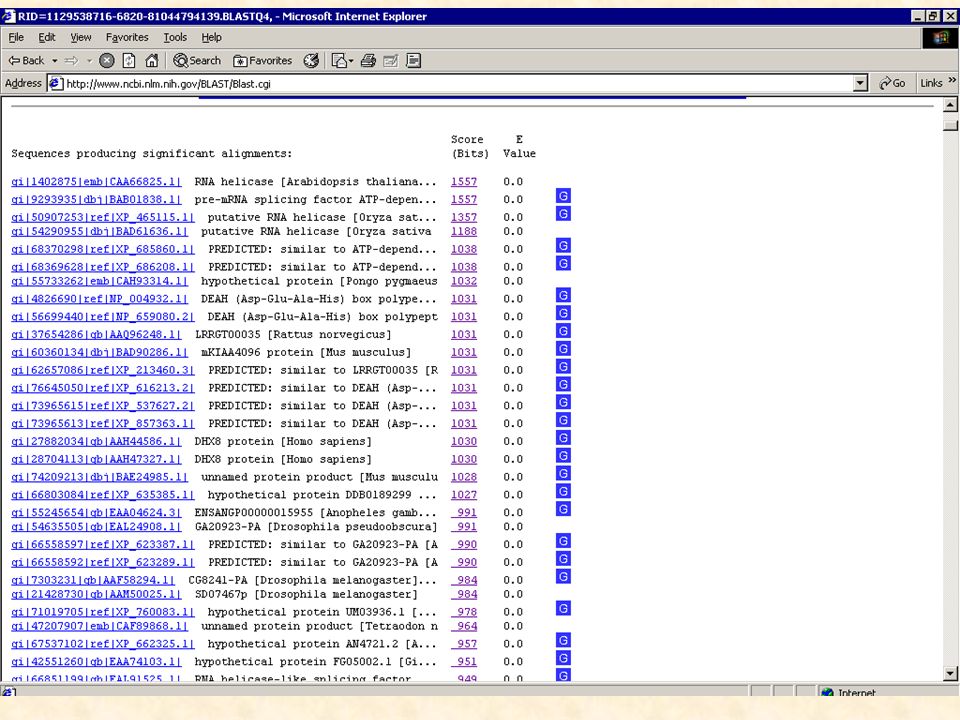
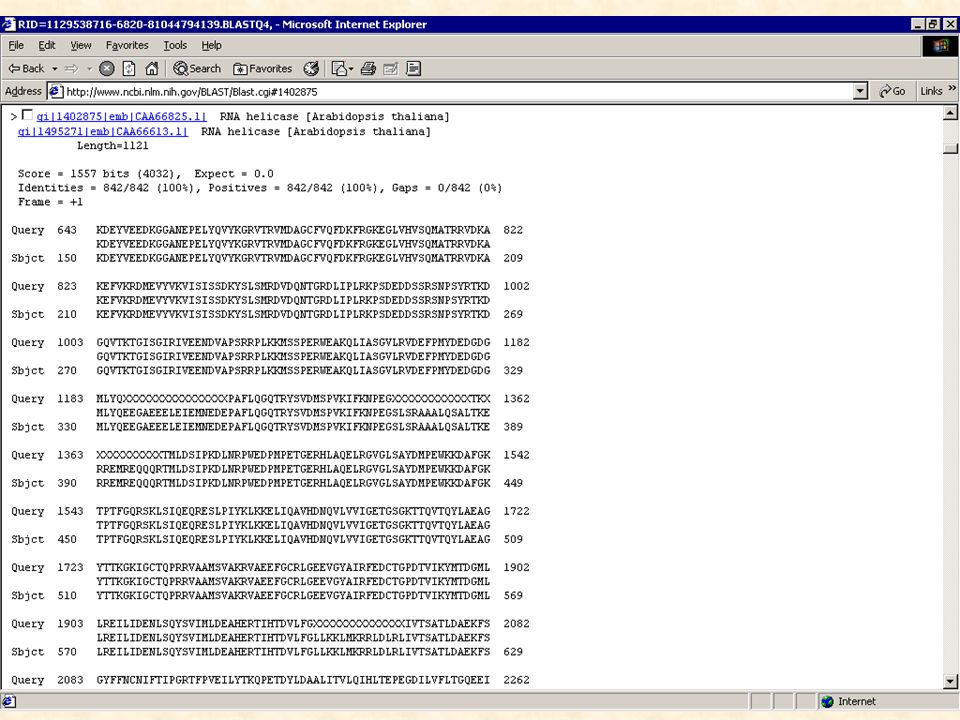
Le logiciel BLAST Basé sur des méthodes statistiques pour déterminer si la similarité observée est significative biologiquement L ’unité fondamentale de BLAST est le HSP (High-scoring Segment Pair) HSP : région de similitude la plus longue possible entre deux séquences ayant un score supérieur ou égal à un score seuil MSP (maximal-scoring Segment Pair) : meilleur score obtenu parmi tous les HSPs que peuvent produire deux séquences 4 programmes distinct de comparaison d ’une séquence avec les bases de données: BLASTN : séquence nucléique contre banque nucléique BLASTP : séquence protéique contre base protéique BLASTX : séquence nucléique traduite en 6 phases contre base protéique TBLASTX : séquence nucléique traduite en 6 phases contre base nucléique traduite sur les 6 phases
 HSP : région de similitude la plus longue possible entre deux séquences ayant. un score supérieur ou égal à un score seuil. MSP (maximal-scoring Segment Pair) : meilleur score obtenu parmi tous les. HSPs que peuvent produire deux séquences. 4 programmes distinct de comparaison d ’une séquence avec les bases de données: BLASTN : séquence nucléique contre banque nucléique. BLASTP : séquence protéique contre base protéique. BLASTX : séquence nucléique traduite en 6 phases contre base protéique. TBLASTX : séquence nucléique traduite en 6 phases contre base nucléique. traduite sur les 6 phases.")
35
L ’algorithme de BLAST La stratégie de la recherche consiste à :
1. Rechercher tous les mots de longueur W dans la séquence W=3 pour les protéines W=11 pour les acides nucléiques 2. Comparer ces mots avec les séquences de la banque afin d’identifier des régions similaires exactes. 3. Extension du segment trouvé dans les deux directions le long de chaque séquence à partir du mot commun pour améliorer le score de l ’alignement. L ’extension s’arrête si: le score descend d ’une quantité x donnée par rapport à la valeur max qu ’il avait atteint le score devient inférieur ou égale à 0 la fin des deux séquences est atteinte

36
blast

37
Résultats de BLAST La signification des alignements est évaluée statistiquement en fonction de : la séquence : longueur et composition la banque de données : taille la matrice utilisée Il appartient au biologiste de déterminer si ces alignements sont significatifs biologiquement ou non.

44
Options du logiciel BLAST
WORLDLENGTH (w) : W correspond au nb de lettres que contiennent les fragments initiaux W=3 pour les protéines et 11 pour les acides nucléiques. Pour augmenter la sensibilité, on peut diminuer la valeur de W mais cela augmente le temps de calcul. FILTER : Cette option, activée par défaut, permet de masquer certaines régions de faible complexité MATRIX : Cette option autorise l ’utilisateur à modifier la matrice utilisée EXPECT : Cette option permet de modifier le score seuil pour la recherche Seuls les alignements dont le score est inférieur à E seront reportés. Plus la valeur de E est faible, plus les résultats obtenus sont pertinents.
 : W correspond au nb de lettres que contiennent les fragments initiaux. W=3 pour les protéines et 11 pour les acides nucléiques. Pour augmenter la sensibilité, on peut diminuer la valeur de W. mais cela augmente le temps de calcul. FILTER : Cette option, activée par défaut, permet de masquer certaines régions. de faible complexité. MATRIX : Cette option autorise l ’utilisateur à modifier la matrice utilisée. EXPECT : Cette option permet de modifier le score seuil pour la recherche. Seuls les alignements dont le score est inférieur à E seront reportés. Plus la valeur de E est faible, plus les résultats obtenus sont pertinents.")
45
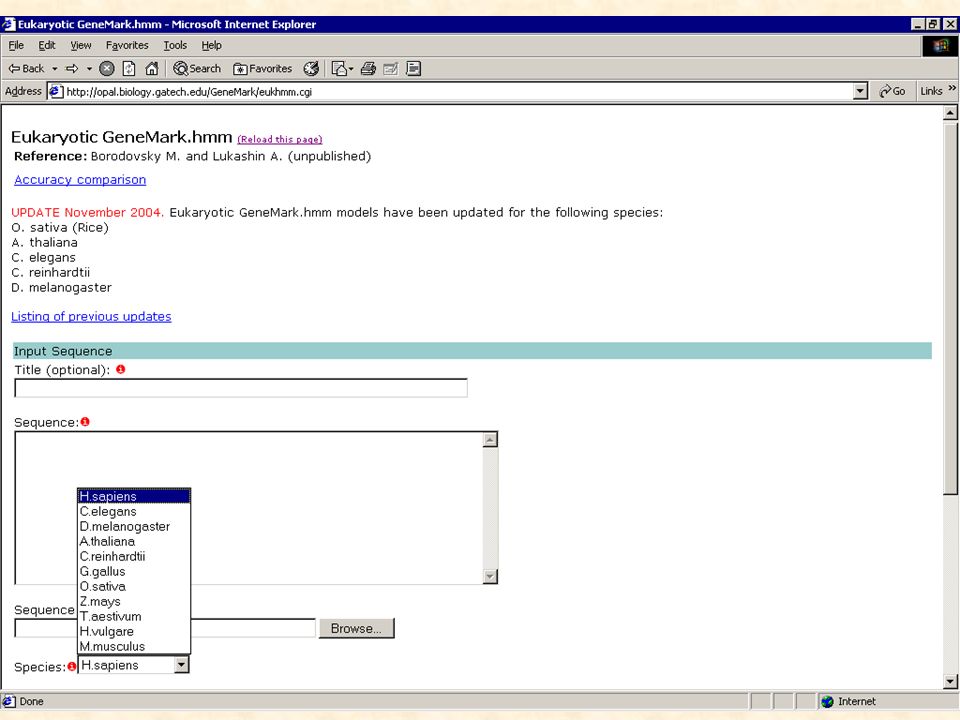
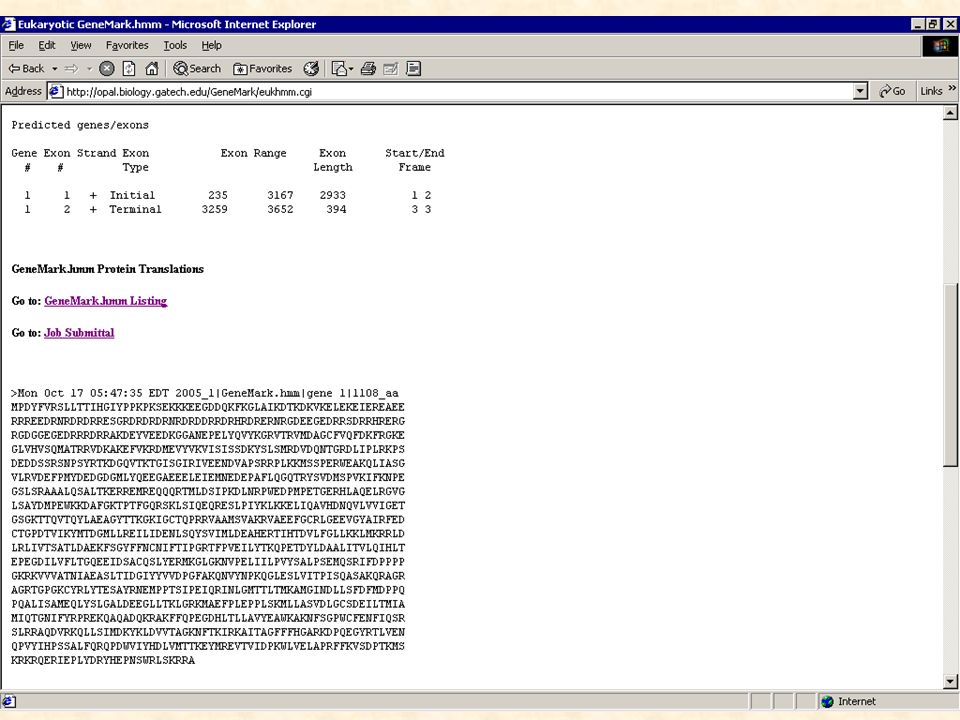
Détection in silico Méthode basée sur des règles consensus concernant : - la détection de signaux de transcription de traduction d’épissage … Elle permet de détecter de nouveaux gènes, ne ressemblant à aucune séquence ou domaine connu.

46
Prédiction de gènes eucaryotes
Les éléments à rechercher sont les suivants: 1. la région promotrice : TATA-box localisée 30 nt en amont du +1 de la transcription motif de Kosac localisé juste en aval du codon ATG initiateur ilots CpG , région riche en dinucléotide CG 2. le signal de polyadénilation : hexamère consensus AATAAA localisé 20 à 30 nt en aval du codon stop 3. les introns sites donneur, accepteur et le nucléotide de branchement

47
Jonctions intron/exon

52
Les erreurs : où et pourquoi ?
Extrémités des gènes sont difficiles à prédire pas ou peu de différence entre les régions intergéniques et les UTR en 3 ’ signaux de polyadénylation sont parfois rares en 5 ’ régions promotrices floues présence d ’exons non codants et d ’introns dans les UTR conséquences : fusion ou cassure des gènes prédits Erreurs internes alignement épissé n’est pas 100% fiable (séquences de mauvaises qualité, vecteurs, polyA) les petits exons sont mal prédits d ’où erreur de structure introns : généralement seuls les sites AG:GT sont recherchés présence d ’événements alternatifs (épissage, initiation traduction)
 les petits exons sont mal prédits d ’où erreur de structure. introns : généralement seuls les sites AG:GT sont recherchés. présence d ’événements alternatifs (épissage, initiation traduction)")
53
Génomique comparative
Approche phylogénétique basée sur la comparaison de deux génomes. => conservation de séquences importantes biologiquement gènes, régions régulatrices

54
Alignement multiple consiste à aligner plusieurs séquences dans leur intégralité : - caractériser des familles de protéines - identifier les régions conservées entre ces séquences - déterminer la séquence consensus de plusieurs séquences alignées trouver, par exemple, un primer consensus pour la PCR … - constituer le point de départ d’une phylogénie Inconvénient: => L ’alignement multiple retourne toujours un résultat Ce traitement n’a de sens que si les séquences à aligner sont supposées proches.

55
Logiciels d’alignement
Clustal X ou W (ftp-igbmc.u-strasbg.fr, ftp.embl-heidelberg.de, ftp.ebi.ac.uk) EXEMPLE FICHIER FORMAT FASTA >CandidaAlbicans SPGRVNLIGDHIDYNFFPWANYFKCALGMKLTFDGNVPTGGG >CandidaParapsilosis SPGRVNLIGDHIDYNYFPWANYFKCGLGMNITFSGTVPTGGGL >YeastGAL1 SPGRVNLIGEHIDYCDFSWSNYFKCGLGLQVFCEGDVPTGSGL
 EXEMPLE FICHIER FORMAT FASTA. >CandidaAlbicans. SPGRVNLIGDHIDYNFFPWANYFKCALGMKLTFDGNVPTGGG. >CandidaParapsilosis. SPGRVNLIGDHIDYNYFPWANYFKCGLGMNITFSGTVPTGGGL. >YeastGAL1. SPGRVNLIGEHIDYCDFSWSNYFKCGLGLQVFCEGDVPTGSGL.")
56
Autres types d’annotations possibles

57
Pol II Annotation des promoteurs
CREB C-myc sp1 +1 transcription TGACGCA CACGTG GGGCGG TATA -25 bp intron exon exon Motifs de fixation des facteurs de transcription sont souvent petits et dégénérés : beaucoup de faux positifs Site d ’initiation de la transcription, n ’est jamais formellement identifié Pas toujours d ’ilots CpG ou de boîte TATA

58
Les gènes non codants Que sont-ils? Gènes produisant des ARN dont la fonction n'est pas de coder pour une protéine. Ces gènes sont transcrits, mais pas traduits. Les ARNt. Potentiellement 64 différents, en pratique une quarantaine dans les génomes microbiens. Probablement beaucoup plus dans les génomes de mammifères. Les ARNr: 5S, 16S, 23S pour les procaryotes, 5.5S, 18S et 28S pour les eucaryotes. Les ARNsn (small nuclear RNA) éléments du spliceosome. Les ARNsno , guides de méthylation.
 éléments du spliceosome. Les ARNsno , guides de méthylation.")
Présentations similaires



>")
















