Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
UE Spécifique ANALYSE ET METHODES D’ ETUDE DU GENOME

2
I- STRUCTURE DU GENOME Organisation Composition
Les différentes séquences Le gène

3
INTRODUCTION Ensemble du matériel génétique d’un organisme
Il est constitué d’ADN pour la majorité des organismes Certains virus ont un génome constitué d‘ARN La taille du génome varie en fonction des espèces

4
ORGANISATION Chez la majorité des bactéries ( procaryotes) le génome est contenu dans UN seul chromosome circulaire Le génome peut être linéaire (actinomycètes) Chez les eucaryotes : ADN nucléaire , ADN mitochondrial
 Chez les eucaryotes : ADN nucléaire , ADN mitochondrial.")
5
DENSITE DES GENOMES Chez E.Coli , le génome est composé presque exclusivement de gènes 4.6 Mégabases / 4000 Gènes / soit 950/MGb Chez l’Homme 3200 Mégabases / Gènes / 8 / MGB La quasi-totalité génome bactérien est codant !

6
Espèce Humaine Génome nucléaire: 3,2 109 pb pour n chromosomes et répartis dans 22 autosomes + 2 chromosomes sexuels Génome mitochondrial : pb, épisome Aucune corrélation entre la taille du génome et la complexité des organismes Plus grands génomes : > pb : pins, plantes

7
COMPOSITION DU GENOME HUMAIN
Des gènes : environ composés de séquences codantes ( exons) et non codantes (les introns, séquences régulatrices; promoteur, silencer, enhancer …) régions codantes : 1.5% du génome ; introns 25%, région régulatrices: 5% Des Pseudogènes 1.5% De très nombreuses séquences répétèes 60% Autres régions non codantes : séquences uniques ou très peu répétées 7%
 et non codantes (les introns, séquences régulatrices; promoteur, silencer, enhancer …) régions codantes : 1.5% du génome ; introns 25%, région régulatrices: 5% Des Pseudogènes 1.5% De très nombreuses séquences répétèes 60% Autres régions non codantes : séquences uniques. ou très peu répétées 7%")
8
Séquences répétées Répétées en Tandem
minisatellites : des séquences de 10 à 25 pb sont répétées un grand nombre de fois ( empreintes génétiques) microsatellites : 1à 5 pb répétées x fois sur plusieurs kb ADN satellite : centromères , télomères ( 10% du génome humain)
 microsatellites : 1à 5 pb répétées x fois sur plusieurs kb. ADN satellite : centromères , télomères ( 10% du génome humain)")
9
Les plus étudiés (CG; CA)n
Microsatellites Dispersés sur tout le génome 1 à 5 nucléotides Copies A(n) / T(n) (CG)n / (CA)n/ (GT)n 106 (CT)n / (GA)n x 106 Les plus étudiés (CG; CA)n CG n = 23 n = 20 n = 15 CGCGCGCG TANDEM
 / T(n) 107. (CG)n / (CA)n/ (GT)n 7 106. (CT)n / (GA)n 3 x 106. Les plus étudiés (CG; CA)n. CG. n = 23. n = 20. n = 15. CGCGCGCG. TANDEM.")
10
Séquences répétées dispersées
Les SINE(s) blocs de 130 à 500 pb ( Alu) Les LINE(s) blocs de 5- 7 kb Les LTR(s) quelques dizaines de paires de bases ( copies) Les transposons ( copies)
 blocs de 130 à 500 pb ( Alu) Les LINE(s) blocs de 5- 7 kb. Les LTR(s) quelques dizaines de paires de bases. ( copies) Les transposons ( copies)")
11
Séquences répétés dispersées
SINEs : Famille ALU Copies : 7 105 pb pb 104 pb ALU ALU Famille Kpn (L1) 1300 pb copies 6 104 Peuvent être transcrites
 1300 pb copies 6 104. Peuvent être transcrites.")
12
Séquences SINEs ALU REPEAT 120 135 290 (AAA)n AT RICH GC RICH GC RICH
n AT RICH GC RICH GC RICH")
13
Eléments transposables
IS éléments 2600 – 700 pb 5' 3' CTGACTT 3' 5' TTCAGTC Répétition aux extrêmités (Maïs)
")
14
Gène Séquence d'acides nucléiques contenant une information codée, transmissible, pour la production régulée d'un ARN (transcription), ce dernier pouvant être traduit en une chaîne polypeptique Certains gènes codent seulement des ARN sans traduction en protéine
, ce dernier pouvant être traduit en une chaîne polypeptique. Certains gènes codent seulement des ARN sans traduction en protéine.")
15
Les gènes des rétrovirus sont constitués d'ARN

16
Pseudogènes Séquences partiellement homologues aux gènes qui ne donnent jamais de protéines correspondantes Soit anciens gènes fonctionnels qui ont muté Soit des ARN retro-transcrit ADN intégration ADN génomique ( transposition)
")
17
ANATOMIE DU GENE b globine

18
1 30 31 104 105 146 IVS1 IVS2 Gène b globine 1600 pb Exon I Exon II
Zone des promoteurs 5' UTR 3' UTR 1 30 31 104 105 146 Exon I IVS1 Exon II IVS2 Exon III Gène b globine 1600 pb

19
Région régulatrice en 5' Zone des promoteurs
Facteurs de transcription -105 -100 -80 -70 CAP CACCC CACCC CCAAT TATA A/T A A/T A G G CCCCC Site d'initiation de la transcription

20
Région 5' non Traduite 5' UTR
CAP Codon initiateur CACCATG +50 CTTCTG Région 5' UTR - Attachement aux ribosomes - Régulation transcription +/-

21
Exon Partie codante d'un gène protéine Exon 1 ATG IVS1 30
Exons Traduction protéine

22
Zone non traduite IVS1 130 pb Exon 2 GT Donneur de l'épissage AG
Accepteur de l'épissage

23
Région 3' UTR Codon 147 146 AATAAA STOP 20 AAAAAA 132 Nucléotides
AATAAA : Signal de polyadénylation : Clivage de l'ARN après transcription (AAAAA)n : Stabilise l'ARN ( ajouté après la transcription)
n : Stabilise l ARN ( ajouté après la transcription)")
24
II ETUDE DU GENOME A- Les expériences fondamentales qui ont révélé l’ existence de l’ADN comme Génome L’ Etude du génome complet d’un individu est très récente et découle du séquençage d’un génome entier ( début XXI)
")
25
C’est l’analyse de l‘ADN
Elle était indirecte avant l’invention des techniques du séquençage (1977) Elle utilisait l’étude des protéines qui renseignaient indirectement sur les gènes Des mutants de bactéries , de drosophiles
 Elle utilisait l’étude des protéines qui renseignaient indirectement sur les gènes. Des mutants de bactéries , de drosophiles.")
26
Origines de l’étude du Génome
Dès le XIX siècle : l’étude de la transmission des caractères sur des critères d’analyses qualitatives et statistiques ( Lois de Mendel 1860) Mise en évidence de l ’ADN (1865) Génétique formelle Morgan ( 1920 drosophile) critères d’analyses qualitatives et statistiques Puis des techniques bactériologiques et biochimiques ont permis de franchir une étape dans le degré d’investigation de l’étude des génomes ( ) Structure ADN (1953 )
 Mise en évidence de l ’ADN (1865) Génétique formelle Morgan ( 1920 drosophile) critères d’analyses qualitatives et statistiques. Puis des techniques bactériologiques et biochimiques ont permis de franchir une étape dans le degré d’investigation de l’étude des génomes ( ) Structure ADN (1953 )")
27
Expérience de Griffith (1928)
Bactérie : Diplococcus pneumoniae 2 souches S = capsule polysaccharidique > infectieux R = dépourvue de capsule > non infectieux

28
Des lots de souris Groupe1 : Injection souche S à des souris > pneumonie Groupe 2 : Injection souche R > pas d’infection Groupe 3 : Injection d’une souche R + souche S préalablement tuée par la chaleur => pneumonie

29
RESULTAT Le sang des souris du groupe 3 est mis en culture : on recueille soit des souches R soit des souches S virulentes ... L’expérience fût refaite en 1944 avec la conclusion que l’ADN était le matériel héréditaire

30
Avery, Mac Leod, Mac Carthy
Interprétation : Les souches S tuées sont capables d’induire une transformation des bactéries R en bactéries S Quelle est la nature de ce matériel transmissible ? En ajoutant l’ ADN purifié des souches S à des colonies de type R les colonies R > S Les autres fractions (polysaccharides,protéines) n’ont pas de pouvoir transformant...
 n’ont pas de pouvoir transformant...")
31
Lorsque l’ADN des souches S est traité par la DNAse avant d’être ajouté aux bactéries de souche R > pas de bactérie de type S La transformation des souches R en souche S est la conséquence d’un transfert d’ADN L’ADN est donc le matériel génétique

32
32

33
De 1944 à 1952 une partie de la communauté scientifique n’était toujours pas convaincue par cette conclusion et l’hypothèse que les protéines étaient le matériel héréditaire était encore défendue

34
Colette VENDRELY ( professeur d’embryologie à Amiens ) & R
Colette VENDRELY ( professeur d’embryologie à Amiens ) & R. VENDRELY démontrent en 1949 que la quantité d‘ADN présente dans les gamètes est la moitié de celle contenue dans les cellules somatiques. Ce résultat est le seul travail français internationalement cité comme contribution majeure à la preuve de l’ADN comme matériel héréditaire
 & R. VENDRELY démontrent en 1949 que la quantité d‘ADN présente dans les gamètes est la moitié de celle contenue dans les cellules somatiques. Ce résultat est le seul travail français internationalement cité comme contribution majeure à la preuve de l’ADN comme matériel héréditaire.")
35
UN pas décisif vers la découverte de la structure de l ’ADN
En 1950 Erwin Chargaff découvre que l'adénine et la thymine existent en quantités égales, et qu'il en est de même de la guanine et de la cytosine, d'où la célèbre équation % A = % T % G = % C

36
Expérience Hershey & Chase
1952, ils démontrent en utilisant le phage T2 traité au tritium * (acides nucléiques radio-actifs) que les ADNs répliqués sont radioactifs. Les protéines ne sont pas radioactives donc ne se répliquent pas …
 que les ADNs répliqués sont radioactifs. Les protéines ne sont pas radioactives donc ne se répliquent pas …")
37
Découverte de la structure de l’ADN 1953

39
Découverte de la structure de l’ADN
39

41
“ It has not escaped our notice that the specific pairing we have postulated immediately suggests a possible copying mechanism for the genetic material ” . « Il n’ a pas échappé à notre attention que la les appariements spécifiques que nous proposons suppose immédiatement un mécanisme de copie du matériel génétique » Le modèle de réplication semi-conservatif sera prouvé en 1958 par Meselson et Stalh

42
Le modèle semi conservatif de la réplication
Le bon modèle pour 3 possibilités

43
modèle conservatif A partir d'une molécule d'ADN bicaténaire "mère", on forme une nouvelle molécule d'ADN bicaténaire. On garde donc ici une molécule "mère", non modifiée (elle est donc conservée), tout en "créant" une nouvelle molécule ("fille").
, tout en créant une nouvelle molécule ( fille ).")
44
On ne conserve aucun brin intact
On ne conserve aucun brin intact. La copie se réalise par fragments dispersés dans l'ensemble de l'ADN, permettant de former les deux molécules d'ADN bicaténaires "filles".

45
Dissociation des deux brins de la molécule d'ADN bicaténaire "mère"
Dissociation des deux brins de la molécule d'ADN bicaténaire "mère". Chaque brin sert donc de matrice à la synthèse d'un brin complémentaire, l'ensemble reformant une molécule d'ADN bicaténaire. Chaque nouvelle molécule "fille" ne conserve donc que la moitié de la molécule mère

46
Dans le tube 0 la totalité de l’ADN est marqué à l’ azote 15
( les 2 brins) . Après une réplication la moitié de l’ADN est radio actif . Au fur et à mesure des réplications la quantité d’ADN marqué diminue l’ADN au profit d’abord d’ ADN hybride , puis d’ADN froid.
 . Après une réplication la moitié de l’ADN est radio actif . Au fur et à mesure des réplications la quantité d’ADN marqué diminue l’ADN au profit d’abord d’ ADN hybride , puis d’ADN froid.")
47
Le résultat observé après séparation des ADNs répliqué correspond au modèle semi conservatif

48
Propriétés physico-chimiques
Déroulement de tout l’ ADN d’une cellule d’un individu : 1.6 Mètre Déroulement de l’ADN de toutes les cellules d’un individu : diamètre du système solaire! ADN cellulaire : 6,6-6,4 x 10 9 pb (2n)
")
49
Quelques valeurs … Pb = Masse Moléculaire = 2x330 g x /6, moles de nucléotides > = 6, g d’ADN Il y a environ 6 picogrammes d’ADN dans une cellule diploide 1 µg d’ADN génomique -> 10-6 g / g/cellule >> 107 cellules diploides

50
II ETUDE DU GENOME B) LES OUTILS
 LES OUTILS")
51
B- 1- PURIFICATION DE l’ ADN Méthode au Phénol / Chloroforme
Purification des cellules Lyse des cellules ( SDS, Lysozyme) Action protéinase K, Rnase Extraction au phénol Action du CHCl3 Précipitation dans l’éthanol en milieu salin Re-suspension en présence de tampon TRIS - EDTA ( 1 mM) ( 100 microG / microl)
 Action protéinase K, Rnase. Extraction au phénol. Action du CHCl3. Précipitation dans l’éthanol en milieu salin. Re-suspension en présence de tampon TRIS - EDTA ( 1 mM) ( 100 microG / microl)")
54
B-2 Les Enzymes de Restriction
Sont des ciseaux moléculaires qui hydrolysent l’ADN; Ils reconnaissent des séquences palindromiques (Ex RADAR) Ils permettent de mette en évidence des variations de séquence ( polymorphismes, mutations) Ils sont utilisés en génie génétique ( clonage) En diagnostic de routine en génétique médicale
 Ils permettent de mette en évidence des variations de séquence ( polymorphismes, mutations) Ils sont utilisés en génie génétique ( clonage) En diagnostic de routine en génétique médicale.")
55
Des enzymes de Restriction

56
5'-G*GATCC-3' Bam HI 3'-CCTAG* G-5’ 5 '-G -3' --- 5'- GATCC-3' 3'-CCTAG-5' ----> 3'- G-5'

58
B-3 L’ Hybridation et les sondes
Hybridation par complémentarité de séquence et appariement des bases Sonde : petite séquence d'ADN ou d'ARN marquée par un composé fluorescent, ou radioactif Les sondes doivent être spécifiques d’une séquence et très sensibles. Utilisation diagnostique

60
S I M P L E B R N Cot (mole sec .l-1) Rapide Lent 100
ADN hautement répétitif S I M P L E B R N % 60 ADN moyennement répétitif ADN peu répétitif 20 10-2 Copies uniques 10-3 100 101 102 104 Cot (mole sec .l-1) Rapide Lent Taux de Renaturation
 Rapide. Lent. Taux de Renaturation.")
61
B-4 Application sondes et ER : "POLYMORPHISME" Plusieurs Formes
Toute variation de séquence de l'ADN, qu'elle soit ponctuelle ou qu'elle intéresse une répétition de mini / micro satellites est un polymorphisme si sa fréquence est 1 % dans une population donnée à une fréquence < 1 % mutation (privée) Les polymorphismes sont soit : - Neutres - Avantageux - Désavantageux
 Les polymorphismes sont soit : - Neutres. - Avantageux. - Désavantageux.")
62
Les Polymorphismes L'ADN n'est pas un élément statique
Il est sujet à des modifications transmissibles Les variations de séquence sont appelées polymorphismes lorsqu'elles surviennent à une fréquence > 0.01 (moins de 1 % : mutations) L'hétérozygotie moyenne de l'ADN humain est d'environ 0,004 (1 base différente toutes les bases entre 2 allèles ou 2 séquences, entre 2 individus)
 L hétérozygotie moyenne de l ADN humain est d environ 0,004 (1 base différente toutes les bases entre 2 allèles ou 2 séquences, entre 2 individus)")
63
Le polymorphisme affecte toutes les régions de l'ADN
- Séquences codantes - Introns - ADN répété (mini, microsat…) Il peut modifier : - 1- Un site de restriction - 2- La longueur d'un motif répété
 Il peut modifier : - 1- Un site de restriction La longueur d un motif répété.")
64
Un polymorphisme peut induire une longueur détectable par enzyme de restriction
Microsatellites Minisatellites

65
Les polymorphismes du DNA servent à son analyse

66
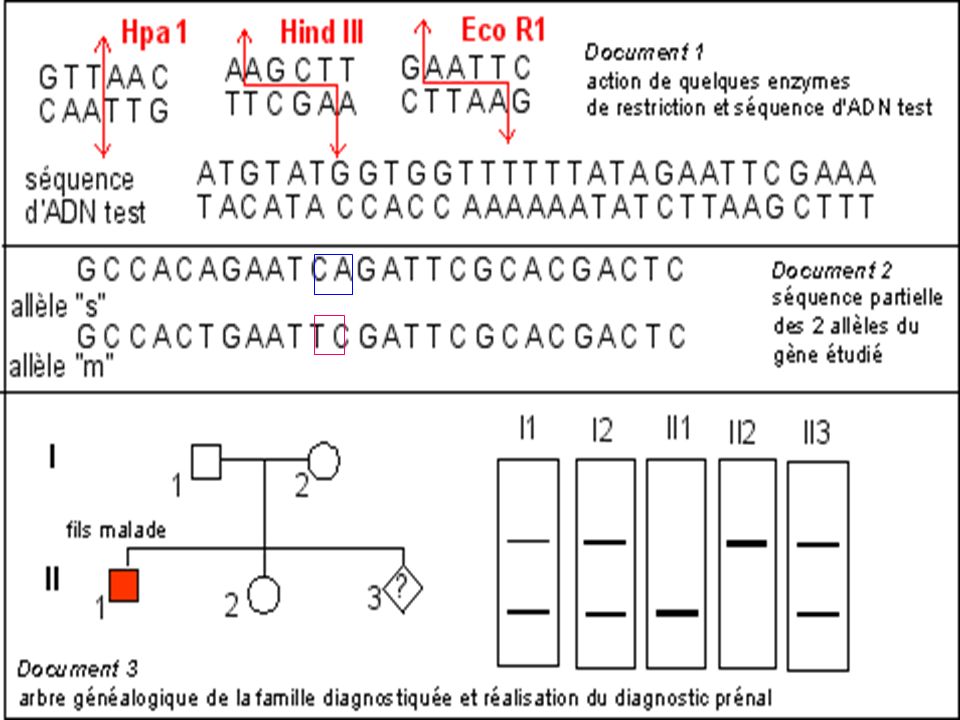
RFLP Un RFLP est défini par un couple : Sonde / Enzyme (Restriction Fragment Lenght Polymorphism) Sa localisation précise, sa variabilité et sa transmission mendélienne lui confèrent un caractère de marqueur génétique codominant. Le couple sonde / enzyme se caractérise par : + / - et correspond à un bi-allélisme Les RFLPs sont mis en évidence par la méthode de Southern ou PCR Pour être informatif : le RFLP doit être reconnu par une sonde unique

67
On distingue des sites de restriction obligatoires (toujours présents) et des sites de restriction variables Polymorphisme Mst II 1 2 3 4 5 20Kb Chez un individu sur 100 on découvre

68
A B Exemple EcoR1 reconnaît, puis hydrolyse 5' GAATTC 3' 3' CTTAAG 5'
Si cette séquence est mutée l'enzyme ECOR1 ne peut plus hydrolyser l'ADN à cet endroit. A B GAGTTC CTCAAG (A + B)
")
69
Ces fragments peuvent être différencier par leur taille
Marqueur Position non connue avec précision Eco RI+ Gène 4kb Eco R1- Gène muté maladie M 5kb Ces fragments peuvent être différencier par leur taille

70
Polymorphisme de Répétition
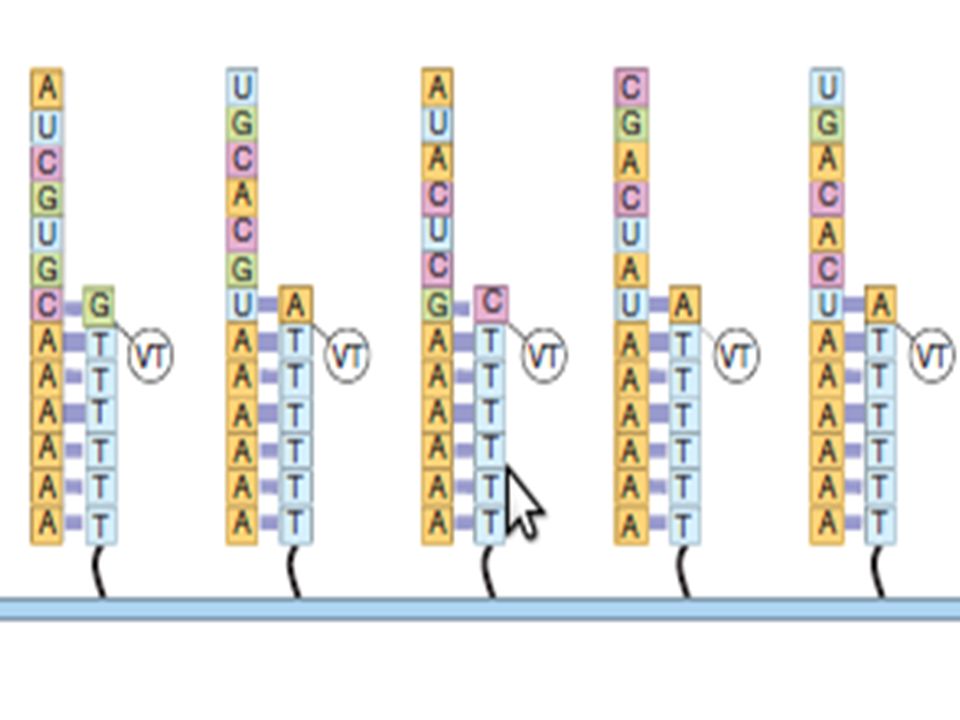
EcoRI EcoRI (CA)n Allèle A 200 pb EcoRI EcoRI + 2(CA)n Allèle B 300 pb sonde A/B A/A B/B 300 200
n. Allèle A. 200 pb. EcoRI. EcoRI. + 2(CA)n. Allèle B. 300 pb. sonde. A/B. A/A. B/B")
71
III- LES TECHNQIUES A - La méthode PCR
Polymerase chain reaction

72
Avantage de la Technique
Elle permet d’étudier un segment d’ ADN au sein de tout le génome si l’on connaît sa séquence Elle permet de recopier ce segment (amplification des millions de fois et de le visualiser par une méthode électrophorétique Par coloration au bromure d’éthidium ou autre colorant Ce segment peut être par la suite traité pour mettre en évidence une mutation Pour le cloner

73
Principe Succession de réactions de réplication d'une matrice double brin d'ADN. Chaque réaction met en oeuvre deux amorces oligonucléotidiques dont les extrémités 3-prime pointent l'une vers l'autre. Les amorces ou «primers» en anglais définissent alors, en la bornant, la séquence à amplifier. Les amorces sont orientées dans le sens 5’ vers 3’

74
Chaque produit de chaque étape de synthèse sert de matrices pour les étapes suivantes. Au lieu d'être linéaire, l'amplification obtenue est exponentielle. Imaginée par K. Mullis en 1985 (Prix Nobel de Chimie dès 1993), la technique connaît un essor considérable à partir de la commercialisation (vers 1988), d'une ADN polymérase résistante aux températures élevées (la Taq polymérase), qui permet une automatisation de la technique en supportant les sauts de température
, la technique connaît un essor considérable à partir de la commercialisation (vers 1988), d une ADN polymérase résistante aux températures élevées (la Taq polymérase), qui permet une automatisation de la technique en supportant les sauts de température.")
75
On opère avec des sauts de température à chaque étape
EN effet, une température dite d’annealing permet l’hybridation de l’amorce avec l’ADN ( 56°) A une autre température , c’est l’ élongation ou extension ( 72 ° C) A une dernière température les brins amplifiés sont séparés : température de dénaturation ( 94°c )
 A une autre température , c’est l’ élongation ou extension ( 72 ° C) A une dernière température les brins amplifiés sont séparés : température de dénaturation. ( 94°c )")
77
Ou Principe de la PCR > ( google)

78
On peut ainsi amplifier assez facilement une région couvrant 2000 pb
Des amplifications de morceaux plus longs sont possibles mais plus délicates ( pb)
")
80
Les délétions mises en évidence par la méthode PCR
B- GAP-PCR / Application de la méthode PCR au diagnostic de grandes délétions Les délétions mises en évidence par la méthode PCR Des amorces choisies pour être très éloignées l’une de l’autre lorsqu’il n’existe pas de délétion , ne permettront pas un amplification En cas de délétion, les amorces sont rapprochées et l’ADN amplifié signe alors une délétion. Dodé C, Krishnamoorthy R, Lamb J, Rochette J. Br J Haematol.1993 Jan;83(1):105-11
:")
81
Distance entre A4 et A9 = 1800 pb : si les gènes alpha 1 et alpha 2 sont présents la
Région amplifiée est de 194pb entre A4 et A1B. Si ,les deux gènes sont délétés la distance entre A4 et A9 est raccourcie et l’amplification révèle un fragment de 550pb.

82
C- LA PCR INVERSE ou Reverse Transcriptase PCR ( RT-PCR)
La RT-PCR a été mise au point pour utiliser les ARN comme matrice d'amplification de la PCR On isole des ARN m par purification spécifique de de ces derniers Ils sont ensuite « recopiés » en ADN par une enzyme : la transcriptase reverse On réalise une PCR comme précédemment Le produit final est un ADN dont l'un des brins est complémentaire de l'ARN d'intérêt et l'autre brin a la même séquence que cet ARN d'intérêt (à la substitution près de U par T).
.")
84
Difficultés Techniques
La contamination des échantillons par de l'ADN génomique est une des principales difficulté de cette technique. En effet, l'ADN génomique peut entrer en compétition avec l'ADNc pour lors de l'étape d'hybridation des amorces. Diverses parades sont alors utilisées :

85
Parades 1 Des ARNm eucaryotes polyadénylés peuvent être purifiés par chromatographie de l'ARN cellulaire total sur une colonne où sont immobilisés des oligo-dT (oligonucléotides constitués de désoxythymidines).
.")
86
2 Les ADN présents dans l'échantillon peuvent être détruits par ajout d'une activité désoxyribonucléase (dépourvue d'activité ribonucléase).
.")
87
3 Choisir les amorces aux bornes d'un intron
3 Choisir les amorces aux bornes d'un intron. Ainsi, les produits d'amplification issus de l'ADNc et ceux issus d'ADN génomique contaminant seront distingués selon leur taille : les fragments issus de l'ADN génomique (contenant l'intron) auront une taille supérieure aux fragments d'amplification issus de l'ADNc (ne contenant pas l'intron).
 auront une taille supérieure aux fragments d amplification issus de l ADNc (ne contenant pas l intron).")
88
D- MUTAGENESE DIRIGEE Réalisation d’une amorce mutée
l'extrémité 3' doit rester complémentaire de la séquence de matrice à amplifier (initiation de la polymérisation) l'extrémité 5' qui peut porter de nombreuses modifications : des mutations ponctuelles, des sites de restriction, des séquences de recombinaison etc.
 l extrémité 5 qui peut porter de nombreuses modifications : des mutations ponctuelles, des sites de restriction, des séquences de recombinaison etc.")
90
Mutation à des extrémités

92
Principe Les amorces 1 et 2 sont des amorces mutées (la région 5' qui porte la mutation est symbolisée par un rectangle rouge). Obtenues par synthèse chimique . Ces amorces sont complémentaires (le recouvrement des régions 5' mutées en rouge est particulièrement important pour la suite). Les amorces 3 et 4 bornent le segment d'ADN à modifier. Elles détermineront la longueur du produit final.
. Obtenues par synthèse chimique . Ces amorces sont complémentaires (le recouvrement des régions 5 mutées en rouge est particulièrement important pour la suite). Les amorces 3 et 4 bornent le segment d ADN à modifier. Elles détermineront la longueur du produit final.")
93
Deux réactions de PCR sont effectuées en parallèle : Une PCR avec les amorces 2 et 3 donne un produit d'amplification correctement muté dans la région désirée, mais de petite taille

95
Une PCR avec les amorces 1 et 4 donne aussi un produit muté, mais toujours de taille insuffisante.

96
Les deux produits de PCR sont purifiés , il s'agit surtout d'éliminer les amorces 1 et 2
On regroupe dans le même tube tous les produits PCR

97
Ils possèdent en commun la région mutée (région d'ADN possédant un rectangle rouge). L'hybridation de ces deux fragments par cette partie commune est critique pour la dernière étape.

98
Une dernière PCR utilisant les amorces 3 et 4 est alors réalisée sur ce mélange et donne, en effet, un fragment de la taille souhaitée contenant la mutation.

100
IV LE SEQUENCAGE ADN ARN

101
Séquençage de l’ADN . F SANGER (1977)
")
102
Séquençage de l’ADN :Walter GILBERT

103
LE SEQUENCAGE PAR LA METHODE DE SANGER
On copie le brin à séquencer de telle sorte que les copies soient radioactives (ou repérables par un autre marquage : fluorescence ) Soit la séquence suivante à déterminer : 3’ …… G C A T G A T C G G 5’ ……Amorce présentera une extrémité 3’ OH libre Il faut choisir une amorce complémentaire d’un bout de séquence connue
 Soit la séquence suivante à déterminer : 3’ …… G C A T G A T C G G 5’ ……Amorce présentera une extrémité 3’ OH libre. Il faut choisir une amorce complémentaire d’un bout de séquence connue.")
105
La DNA polymérase réplique 5' 3' à partir 3'OH libre de l’amorce
en présence d'un 2, 3 didéoxynucléotide la réplication est stoppée. Statistiquement on observera des fragments d'ADN de tailles différentes en fonction de l'incorporation du 2, 3 didéoxynucléotide pendant la réplication du brin à séquencer

106
Tube 1 4dNTP, ddGTP 3' GCATGATCGG CG# CGTACTAG# Tube 2 4dNTP, ddATP 3'GCATGATCGG CGTA# CGTACTA#

107
Tube 3 4dNTP, ddCTP 3' GCATGATCGG 5' ddC # CGTAC# CGTACTAGC# CGTACTAGCC# Tube 4 4dNTP, ddTTP CGT# CGTACT#

108
Tube 1 : ddGTP 1er fragment : 2 bases 2nd fragment : 8 bases Tube 2 : ddATP 1er fragment : 4 bases 2nd fragment : 7 bases Tube 3 : ddCTP 1er fragment : 1 base 2nd fragment : 5 bases 3ème fragment : 9 bases 4ème fragment : 10 bases Tube 4 : ddTTP 1er fragment : 3 bases 2nd fragment : 6 bases

109
Remarques : Il n'y a jamais deux fois la même taille quelque soit le tube examiné Les produits sont radioactifs, ils vont être repérés par autoradiographie après électrophorèse en gel d'acrylamide. Les fragments migreront d'autant plus vite que leur masse moléculaire est faible. La lecture se fera de 5’ vers 3 ‘ en commençant par les fragments les plus petits ( du bas vers le haut)
")
110
On lit donc Lecture : 5' CGTACTAGCC 3' Séquence : 3' GCATGATCGG 5‘ On donne le produit complémentaire du produit de lecture : 3’ …… G C A T G A T C G G 5’

112
Sanger Centre-Cambridge

113
Sequençage direct de l’ARN
ON ne transforme pas en cDNA Des séquences de poly T sont bloquées sur un support Le support capture les ARN par leur extrémité poly A 3’dA : 3’ Desoxy A bloque la fin de séquence d’ARN Il sont ensuite complétés avec addition de Thymidine non marquée Puis bloqués avec des nucléotides fluorescents A et C et G , dit terminateurs + Polymérase. Cette méthode permet le début d’une lecture par fluorescence là où commence le RNA et ne lit pas le poly A .

116
V- ANALYSE DE l’ADN Par les méthodes de séparation de fragments

117
V-A Techniques Electrophorétiques
La Séparation des fragments d’acides nucléiques V-A Techniques Electrophorétiques Les macromolécules chargées électriquement peuvent migrer dans un champ électrique. La densité de charge par unité de longueur est la même dans l’ARN et l’ADN (contrairement aux protéines). La migration s’effectue du Pôle - vers le Pôle + du générateur. La différence de migration se fera donc par la taille Le support de migration ( acrylamide, agarose) jouant un rôle de filtre, les fragments les plus petits migreront plus rapidement que d’autres plus longs dans le dispositif classique.
. La migration s’effectue du Pôle - vers le Pôle + du générateur. La différence de migration se fera donc par la taille. Le support de migration ( acrylamide, agarose) jouant un rôle de filtre, les fragments les plus petits migreront plus rapidement que d’autres plus longs dans le dispositif classique.")
119
Coloration au Bromure d’éthidium sous lampe UV

120
La migration s’effectue du haut vers bas
La migration s’effectue du haut vers bas . Les fragments les plus petits sont en bas du gel de migration

122
Électrophorèse d’ADN humain sans hydrolyse par ER : Smear

123
V-B LA METHODE DE SOUTHERN

124
SOUTHERN BLOT 1) Extraction de l'ADN des différents génotypes à analyser 2) Digestion de l'ADN par un enzyme de restriction. On obtient alors des fragments d'ADN de longueurs différentes selon les individus. Cette différence de taille est due a une différence du nombre de site de restriction qui varie selon les individus. l'ADN à analyser est fragmenté en séquences plus ou moins longues en fonction du polymorphisme de longueur des fragments de restriction.
 Digestion de l ADN par un enzyme de restriction. On obtient alors des fragments d ADN de longueurs différentes selon les individus. Cette différence de taille est due a une différence du nombre de site de restriction qui varie selon les individus. l ADN à analyser est fragmenté en séquences plus ou moins longues en fonction du polymorphisme de longueur des fragments de restriction.")
125
3) Ces fragments de restriction sont séparés selon leur taille par une électrophorèse en gel d'agarose. L'ADN étant chargé négativement, il migre de la cathode vers l'anode. Les fragments les plus petits sont les plus rapides. 4) LE BLOT / L'ADN est ensuite transféré sous forme dénaturée ( NAOH, KOH, 1M) sur une membrane de nylon. La position relative des fragments d'ADN est préservée durant le transfert.
 LE BLOT / L ADN est ensuite transféré sous forme dénaturée ( NAOH, KOH, 1M) sur une membrane de nylon. La position relative des fragments d ADN est préservée durant le transfert.")
126
5 ) L’ HYBRIDATION : La membrane est incubée dans une solution contenant une sonde marquée préalablement, soit par la radioactivité, soit chimiquement. La sonde s'hybride alors avec le ou les fragments d'ADN avec lesquels elle présente une homologie. On utilise couramment deux types de sondes : les sondes génomiques (ADNg) et les sondes d'ADN complémentaire (ADNc).
 et les sondes d ADN complémentaire (ADNc).")
127
Les sondes génomiques sont obtenues par digestion de l'ADN total du génome nucléaire de l'espèce étudiée à l'aide d'un enzyme de restriction. Les sondes d'ADNc correspondent nécessairement à des gènes exprimés, puisqu'elles sont obtenues à partir des ARN messagers.

128
6) L'endroit, ou les endroits, où la sonde s'est fixée sont révélés en plaçant la membrane au contact d'un film sensible à la radioactivité (figure), ou en réalisant une réaction enzymatique colorée spécifique..
 L endroit, ou les endroits, où la sonde s est fixée sont révélés en plaçant la membrane au contact d un film sensible à la radioactivité (figure), ou en réalisant une réaction enzymatique colorée spécifique..")
129
4.2Kb 4kb 2Kb

130
Interprétation des résultats considérant 2 allèles
Sites Eco R1 4Kb 2Kb Sonde 4.2kb Site polymorphe sur le 2 ième allèle :

131
Interprétation du résultat
Des individus sont 4/2 // 4/ 2 (homozygotes) Des Individus ne montrent aucune hybridation -- Délétion d’au moins 6 kb (homozygote) Le dernier : 4/2 // 4.2/2 : ses deux allèles sont différents ( hétérozygote)
 Des Individus ne montrent aucune hybridation. -- Délétion d’au moins 6 kb (homozygote) Le dernier : 4/2 // 4.2/2 : ses deux allèles sont différents ( hétérozygote)")
133
Diagnostic de la drépanocytose
Maladie génétique fréquente en Afrique noire au Moyen Orient et en Inde . Maladie récessive Mutation ponctuelle par substitution Glu > Val sur le gène beta globine ( codon 6) GAG > GTG Cette mutation supprime un site de restriction pour l’enzyme Mst II
 GAG > GTG. Cette mutation supprime un site de restriction pour l’enzyme Mst II.")
136
LE NORTHERN BLOT // Southern BLOT Isolement de l’ARN colonne oligo dT
Electrophorèse sans dénaturation , sans hydrolyse Hybridation avec une sonde monobrin

137
LA GENOMIQUE Science des Génomes
La génomique regroupe un ensemble d'analyses qui vont : - d’une cartographie physique de l’ADN - d’ une cartographie des ARN messagers - à l'identification de gènes , de polymorphismes et de séquences d’intérêts - à l'étude de leurs fonctions

138
Parler de brin sens ou antisens sur l'ADN n'a de sens (humour) que si nous nous plaçons dans le contexte de la transcription. Le brin sens est celui qui a la même séquence nucléotidique que l'ARN messager transcrit (T / U mis a part bien sûr). Le brin antisens sert de matrice à la polymerase. L'ARN est polymerisé de 5' vers 3'. La matrice (brin "antisens") peut donc être représenté du 3' au 5' Le brin complémentaire (brin "sens") est donc représenté du 5' au 3'. sens ’ -ATTTGCTGCCTT... TGC-3' antisens 3'-TAAACGACGGAA.. ACG-5' RNA messager : 5'-AUUUGCUGCCUU..UGC-3’ La lecture d’une séquence par la méthode de Sanger lit (en commençant par la talile la plus petite ) de 5’ vers 3’ c’est à dire le brin sens
 de 5’ vers 3’ c’est à dire le brin sens.")
139
Cartographie de l’expression des GENES
Hybridation de l’ARN sur puces à ADN

140
Analyse totale du génome par utilisation de
l’expression différentielle des gènes On construit des supports ou chip-arrays . Chaque grille est un carré de 11 micromètres porteur de séquences d’ ADN (oligoprobe ( complémentaire à celle de séquences de gènes) Des Millions de brins de DNA gréffés et accessibles 6.5 millions de brin sur chaque chip Brins = 25 base pairs
 Des Millions de brins de DNA gréffés et accessibles. 6.5 millions de brin sur chaque chip. Brins = 25 base pairs.")
142
Chaque espèce d’ARN va hybrider avec la sonde d’ADN correspondante sur la puce
préalablment chargée de sondes d’ADN. La combinaison ARN biotinylé – ADN est fluorescente et donc repérable par un système de lecture approprié Fluorescent Stain Biotin RNA After staining, RNA (purple) bound to the DNA probe built on the array will fluoresce RNA (purple) has bound to its DNA probe built on the array
 bound to the DNA probe built on the array will fluoresce. RNA (purple) has bound to its DNA probe built on the array.")
Présentations similaires






















>")


 Obtention de l’ADN recombinant>")
 Obtention de l’ADN recombinant>")
 polymorphes (entre individus, espèces, …) permettant - l’établissement de cartes.>")






 Phosphatases: retirent un phosphate.>")





