Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
« Genetic Suppression of Polyglutamine Toxicity in Drosophila »
DROSOPHILA AS MODEL FOR HUMAN NEURODGENERATIVE DISEASES « Expanded Polyglutamine Protein Forms Nuclear Inclusions and Causes Neural Degeneration in Drosophila » John M. Warrick et al (1998) Cell 93, 939–949 « Genetic Suppression of Polyglutamine Toxicity in Drosophila » Parsa Kazemi-Esfarjani and Seymour Benzer (2000) Science 287 :
 Cell 93, 939–949. « Genetic Suppression of Polyglutamine Toxicity in Drosophila » Parsa Kazemi-Esfarjani and Seymour Benzer (2000) Science 287 :")
2
Maladie d’Alzheimer: en France, prévalence de 1 % entre 65 et 69 ans à plus de 15 % au delà des 85 ans nouveaux cas /an de maladie d'Alzheimer dont environ les 2/3 survenant chez des personnes de plus de 79 ans. Estimation 2010: cas. Atrophie cérébrale généralisée progressive présénile entraînant l'apparition d'une démence lentement évolutive avec >Troubles de langage jusqu’à l’ aphasie (perte de la parole) >Troubles cognitifs: troubles de mémoire, exécutifs : planification d’actions >Agnosie (déficit de la capacité de reconnaissance ) >Apraxie (incapacité à effectuer un mouvement ou une série de mouvements sur consigne, trouble de coordination des mouvements) » > Perte d’autonomie progressive (perte capacité à assurer ses besoins quotidiens) Maladie de Huntington :prévalence 3 à 7/ en europe de l’ouest Début tardif : ans ( ans), 6% de moins de 20 ans (transmission paternelle) >Mouvements anormaux involontaires (Mouvements choréoathétosiques: mouvements saccadés , explosifs, imprévisibles), Troubles de la marche : démarche raide, petits pas >Troubles cognitifs: troubles de mémoire et d’attention suivis de trouble du langage (dysarthrie) et des gestes >Troubles psychiatriques: dépression (50%); troubles de caractère, psychoses (10%). > Perte d’autonomie progressive
 >Troubles cognitifs: troubles de mémoire, exécutifs : planification d’actions. >Agnosie (déficit de la capacité de reconnaissance ) >Apraxie (incapacité à effectuer un mouvement ou une série de mouvements sur consigne, trouble de coordination des mouvements) » > Perte d’autonomie progressive (perte capacité à assurer ses besoins quotidiens) Maladie de Huntington :prévalence 3 à 7/ en europe de l’ouest. Début tardif : ans ( ans), 6% de moins de 20 ans (transmission paternelle) >Mouvements anormaux involontaires (Mouvements choréoathétosiques: mouvements saccadés , explosifs, imprévisibles), Troubles de la marche : démarche raide, petits pas. >Troubles cognitifs: troubles de mémoire et d’attention suivis de trouble du langage (dysarthrie) et des gestes. >Troubles psychiatriques: dépression (50%); troubles de caractère, psychoses (10%). > Perte d’autonomie progressive.")
3
Maladie de Machado-Joseph (ataxie spinocérébelleuse de type 3 ,SCA3):
Dégénérescence spinocérébelleuse (de la moelle épinière et du cervelet) 3 types sont décrits La forme la plus courante est de Type 2 appelé type ataxique : Troubles de la démarche et ataxie des extrémités se déclarant à la deuxième et la quatrième décennie Ataxie cérébelleuse (manque de coordination fine des mouvements volontaires): Dysarthrie (difficulté à articuler les mots) Mouvements avec une amplitude trop importante (le patient semble être ivre) Impossibilité d'exécuter des mouvements rapides Tremblements des membres d'un seul côté au cours des mouvements volontaires (atteinte de la moitié du cervelet) Syndrome extrapyramidal : Tremblement régulier qui atteint généralement l'extrémité des membres supérieurs. Le tremblement s’accentue quand le patient se concentre en particulier en effectuant un calcul mental et disparaît quand le patient effectue des mouvements sous le contrôle de la volonté. Akinésie (lenteur d'initiation des mouvements avec une tendance à l'immobilité ) Clignements des paupières de plus en plus rares associés à une mimique appauvrie. Perte du balancement des bras au moment de la marche qui s'effectue à petits pas. Hypertonie des membres qui donne au patient une attitude fléchie, penchée en avant. Difficultés dans l'exécution des mouvements alternatifs rapides Economie des gestes Ecriture micrographique (lettres minuscules) Paroles exprimées sur le même ton Tremblements de la langue et du visage Ophtalmoparésie (paralysie légère consistant en une diminution des possibilités de contraction des muscles du globe oculaire).
 3 types sont décrits. La forme la plus courante est de Type 2 appelé type ataxique : Troubles de la démarche et ataxie des extrémités se déclarant à la deuxième et la quatrième décennie. Ataxie cérébelleuse (manque de coordination fine des mouvements volontaires): Dysarthrie (difficulté à articuler les mots) Mouvements avec une amplitude trop importante (le patient semble être ivre) Impossibilité d exécuter des mouvements rapides. Tremblements des membres d un seul côté au cours des mouvements volontaires (atteinte de la moitié du cervelet) Syndrome extrapyramidal : Tremblement régulier qui atteint généralement l extrémité des membres supérieurs. Le tremblement s’accentue quand le patient se concentre en particulier en effectuant un calcul mental et disparaît quand le patient effectue des mouvements sous le contrôle de la volonté. Akinésie (lenteur d initiation des mouvements avec une tendance à l immobilité ) Clignements des paupières de plus en plus rares associés à une mimique appauvrie. Perte du balancement des bras au moment de la marche qui s effectue à petits pas. Hypertonie des membres qui donne au patient une attitude fléchie, penchée en avant. Difficultés dans l exécution des mouvements alternatifs rapides. Economie des gestes. Ecriture micrographique (lettres minuscules) Paroles exprimées sur le même ton. Tremblements de la langue et du visage. Ophtalmoparésie (paralysie légère consistant en une diminution des possibilités de contraction des muscles du globe oculaire).")
4
Number of common features characterized human neurodegenerative diseases:
-Late–onset and progressive neurodegeneration -Dysfunction and loss of specific neurons in the brain -Formation of abnormal protein aggregates intranuclear inclusions : Huntington’s disease and related polyglutamine disorders cytoplasmic inclusions : neurofibrillary tangle of Alzheimer’s disease , Lewy body of Parkinson’s disease. extracellular : amyloid plaque of Alzheimer’s disease

5
At least nine human neurodegenerative diseases known as polyglutamine (polyQ) diseases
has been defined as CAG repeat expansion within the ORF of the respective genes HD Huntington’s Disease DRPLA Dentatobulbar Muscular Atrophy SCA Spinocerebellar Ataxia (also known as MJD : Machado Joseph Disease), SCA 17 . SBMA Spinobulbar Muscular (also known as Kennedy disease)
, SCA 17 . SBMA Spinobulbar Muscular (also known as Kennedy disease)")
6
Study of the molecular and cellular mechanisms underlying
the disease in transgenic animals Why the fly? - Short life span of days, large number of progeny - Small/ simple (non-redundant) genome - High conservation of fundamental biological pathways - 75% of human disease genes have a Drosophila ortholog - Many tools to manipulate gene expression Easy forward and reverse genetic approaches Large scale pharmacological screens possible Fly nervous system: - Anatomy and development of drosphila nervous system characterized - Tools to identify specific neuron subtype available - Measurement of Neuronal function and survival , learning , memory
 genome. - High conservation of fundamental biological pathways. - 75% of human disease genes have a Drosophila ortholog. - Many tools to manipulate gene expression. Easy forward and reverse genetic approaches. Large scale pharmacological screens possible. Fly nervous system: - Anatomy and development of drosphila nervous system characterized. - Tools to identify specific neuron subtype available. - Measurement of Neuronal function and survival , learning , memory.")
7
SCA3 /MDJ3 ATAXIN 3 (histone binding protein , ubiquitin protease domain)
Truncated forms with normal or expansion of polyQ (Q27 and Q78) of the human protein mis-expressed using the UAS –GAL4 system
 of the human protein mis-expressed using the UAS –GAL4 system.")
8
UAS-MJDtr-Q27 (Contrôle) et UAS-MJDtr-Q78
Hsp 70 12 aa (CAG n) 43 aa Hemagglutinin (HA) tag COOH terminal fragment of Ataxin 3 (Loss of integrity nervous system)
 43 aa. Hemagglutinin (HA) tag. COOH terminal fragment of Ataxin 3. (Loss of integrity nervous system)")
9
Scanning electron photomicrograph
The Drosophila compound eye is a complex neural tissue with precise cellular architecture. The adult eye is composed of 800 reiterated subunits called ommatidia. Tangential section

10
8 photoreceptors and 8 non-neural support cells arranged in an invariant pattern.
R1 Photoreceptor cell R1 Photoreceptor cell rhabdomere (light-sensing organelle, contain rhodopsin)
")
11
The morphogenetic furrow (mf) marks the passage of a wave of cellular differentiation
anterior posterior Third instar eye-antennal imaginal disc, labeled with an antibody which recognizes all the photoreceptor neurons (R1-R8) Cellular differentiation initiates during the middle of the third larval instar stage at the posterior edge of the eye disc and sweeps across in an anterior direction, taking approximately 2 days to traverse the entire field eye antenna Gradient of photoreceptor recruitment and differentiation
 Cellular differentiation initiates during the middle of the third larval instar stage at the posterior edge of the eye disc and sweeps across in an anterior direction, taking approximately 2 days to traverse the entire field. eye. antenna. Gradient of photoreceptor recruitment and differentiation.")
12
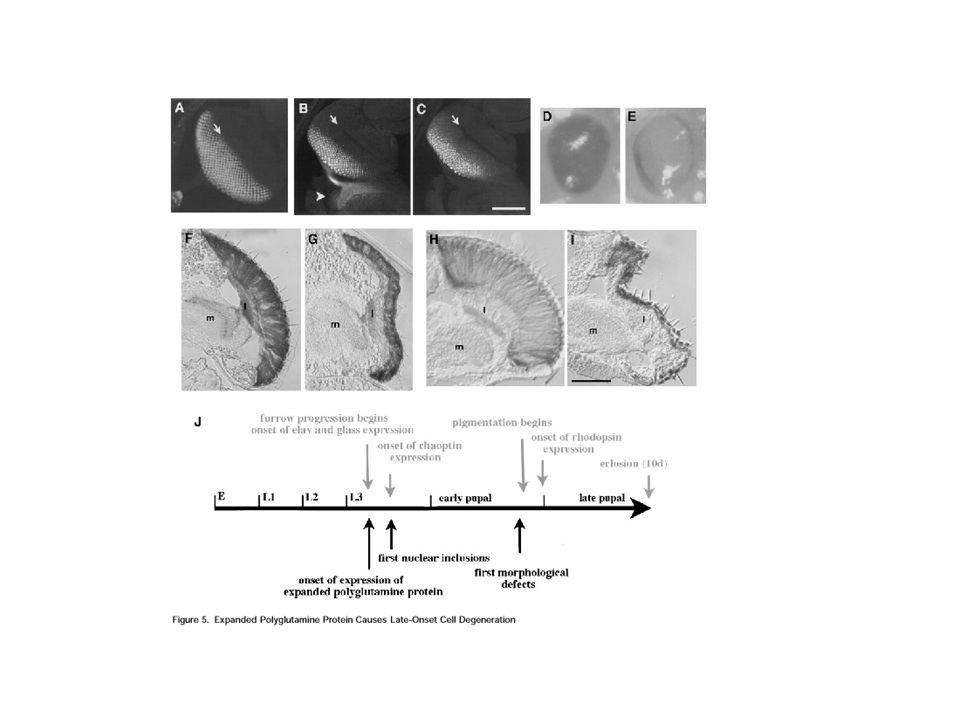
FIG 1 Expanded polyglutamine protein causes adult eye degeneration
Neurones du système nerveux central et périphérique durant tout le développement Elav-GAL4 UAS-Q78 (M)/+ Elav-GAL4 UAS-Q27/+ 1 day old 4 day old 4 day old Progressive Loss of photoreceptor morphology 1 day old 7day old 7 day old Progressive Pigment degeneration gmr-GAL4 UAS-Q78 (W)/+ gmr-GAL4 UAS-Q27/+ Toutes les cellules de l’œil en cours de développement ( cellules photoreceptrices et cellules pigmentaires)
/+ Elav-GAL4 UAS-Q27/+ 1 day old. 4 day old. 4 day old. Progressive Loss of photoreceptor morphology. 1 day old. 7day old. 7 day old. Progressive Pigment degeneration. gmr-GAL4 UAS-Q78 (W)/+ gmr-GAL4 UAS-Q27/+ Toutes les cellules de l’œil en cours de développement. ( cellules photoreceptrices et cellules pigmentaires)")
13
Moderate Strong GMR-GAL4 UAS-Q78 UAS-Q27 Weak GMR-GAL4 Age: 1 jour
all cells in the developping eye Pigmentation Structure externe Section Frontale Cerveau Dégenerescence cellulaire niveau rétine (r) Eye and brain morphology of flies expressing MDJtrQ78 suggest a late stage loss of photoreceptor integrity
 Eye and brain morphology of flies expressing MDJtrQ78 suggest a late stage loss of photoreceptor integrity.")
14
Moderate Strong Immunofluorescence sur disque imaginal oeil UAS-Q78
Protéines MJDtr révélées grâce à l’anticorps anti HA UAS-Q78 UAS-Q27 GMR-GAL4 Weak Moderate Strong MJDtr Q27cytoplasmique MJDtr Q78 inclusions nucléaires Différenciation séquentielle des omatidies postérieurement au sillon morphogénétique

15
anti HA Chromatine anti HA ; Chromatine GMR-GAL4 UAS Q27 GMR-GAL4
UAS Q78 (S) anti HA ; Chromatine anti HA; Membrane nucléaire
 anti HA ; Chromatine. anti HA; Membrane nucléaire.")
16
Figure 4: NI formation in various Imaginal tissues
dpp-GAL4/ UAS-Q78(S) dpp-GAL4/ UAS-Q27 Anti HA Chromatin dpp-GAL4/ UAS-Q78(S) NI formation but no phenotype >> NI not sufficient to cause degeneration in vivo
 dpp-GAL4/ UAS-Q27. Anti HA. Chromatin. dpp-GAL4/ UAS-Q78(S) NI formation but no phenotype >> NI not sufficient to cause degeneration in vivo.")
18
GAL 4 UAS Hsp 70 20 ou 127 CAGs hemagglutin (HA)- tag GMR GAL4
Eye specific UAS 20Q UAS 127 Q UAS Hsp 70 20 ou 127 CAGs hemagglutin (HA)- tag
- tag.")
19
Target P element: EP Gal4 UAS Hsp 70 20 ou 127 CAGs

20
HA tag HA tag chromatine NI

23
dTRP2 interaction with the drosophila homolog of NF1 (dNF1)
TPR2 interact with the GTPase–activating protein-related domain (GRD) of Neurofibromin (NF1 gene product) The NF1 GRD accelerates intrinsic activity of Ras-GTPase, resulting an active Ras-GTP to be converted into an inactive GDP form Hypothesis : dTRP2 interaction with the drosophila homolog of NF1 (dNF1) increase Ras-GTP activity >>inhibit the proapoptotichead involution defective (HID) protein enhance the survival of eye cells.
 of Neurofibromin. (NF1 gene product) The NF1 GRD accelerates intrinsic activity of Ras-GTPase, resulting an active Ras-GTP to be converted into an inactive GDP form. Hypothesis : dTRP2 interaction with the drosophila homolog of NF1 (dNF1) increase Ras-GTP activity. >>inhibit the proapoptotichead involution defective (HID) protein. enhance the survival of eye cells.")
24
dTPR2 and dHdJ1 (HSP40/HdJ1) contain J domain (stimulates ATPase activity of HSP70)
Molecular chaperone may play a role in disease progression Nuclear Inclusion immunostain for ubiquitin. May contain misfolded or abnormally folded protein potentially targeted for proteasomal degradation. Nuclear Inclusion immunostain for HSP70.

25
Supression of toxicity :
restauration external and internal eye structure by overexpression in MdJDtr-Q78 (A) of : -HSP70 (B) -dHdJ1 (C) -HSP70 + dHdJ1(D) In supressed flies: NI still present and morphology/number/size unchanged Pathogenic protein is SDS-soluble and detected as a monomeric protein by Western immunoblot Hypothesis: chaperones are modulating the structure of the protein, and this is not visible in the large aggregates chaperones, by interacting with the disease protein, prevent abnormal interactions with other proteins in the cell that are causal in toxicity - pathogenic disease proteins may cause a slow cellular depletion of chaperone activity leading to the failure of many cellular processes. Bonini (2002) PNAS 99: 16407–16411
 of : -HSP70 (B) -dHdJ1 (C) -HSP70 + dHdJ1(D) In supressed flies: NI still present and morphology/number/size unchanged. Pathogenic protein is SDS-soluble and detected as a monomeric protein by Western immunoblot. Hypothesis: chaperones are modulating the structure of the protein, and this is not visible in the large aggregates. chaperones, by interacting with the disease protein, prevent abnormal interactions with other proteins in the cell that are causal in toxicity. - pathogenic disease proteins may cause a slow cellular depletion of chaperone activity leading to the failure of many cellular processes. Bonini (2002) PNAS 99: 16407–")
28
GMR -GAL4 - + +

Présentations similaires




 Analysis of instruments and actions to support eco-innovation and eco-investment.>")












 mean-field (as a general theory)>")



>")

