Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Analyse statistique des séquences génomiques
DEA en bioinformatique Marseille 10 Novembre 2000 Laurent Duret

2
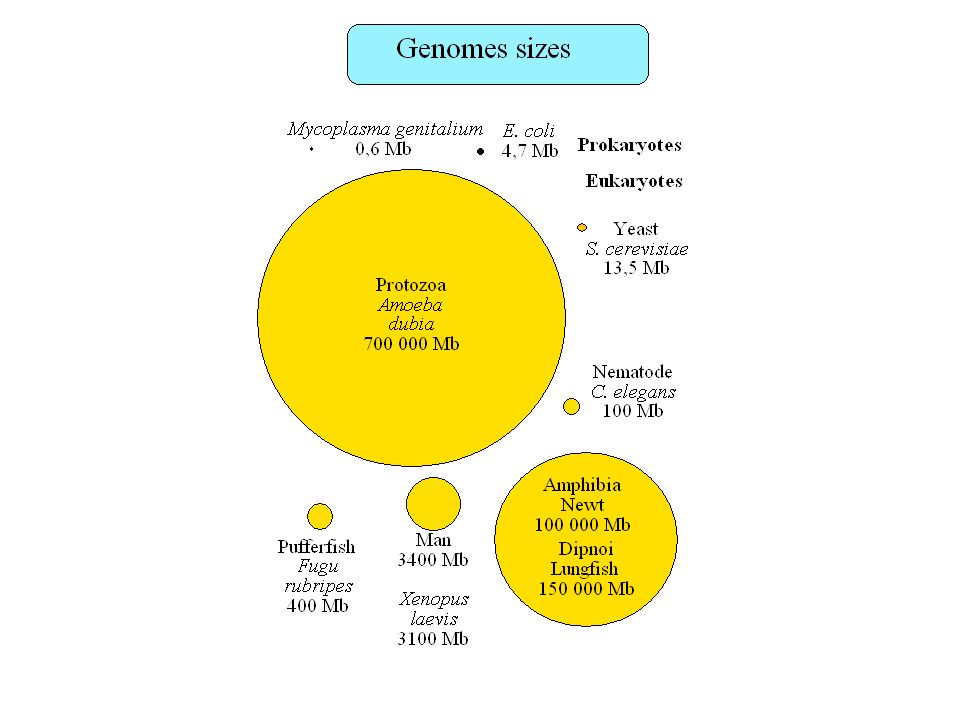
Plan Taille des génomes, paradoxe de la valeur C
Contenu informationnel Séquences répétées Organisation en isochore des génomes de vertébrés Projets génomes: état des lieux Recherche de gènes protéiques Méthodes ab initio Utilisation des EST Approche comparative Recherche de régions régulatrices non-codantes

7
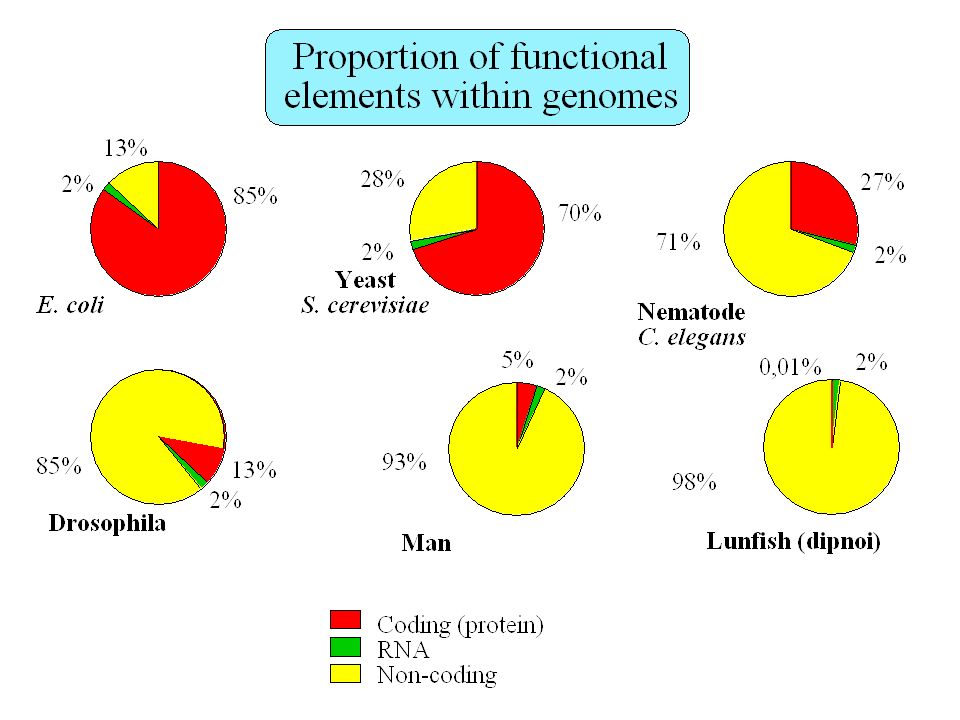
Functional elements in the human genome
Untranslated RNAs: Xist, H19, His-1, bic, etc. Regulatory elements: promoters, enhancers, etc. 2% ??

8
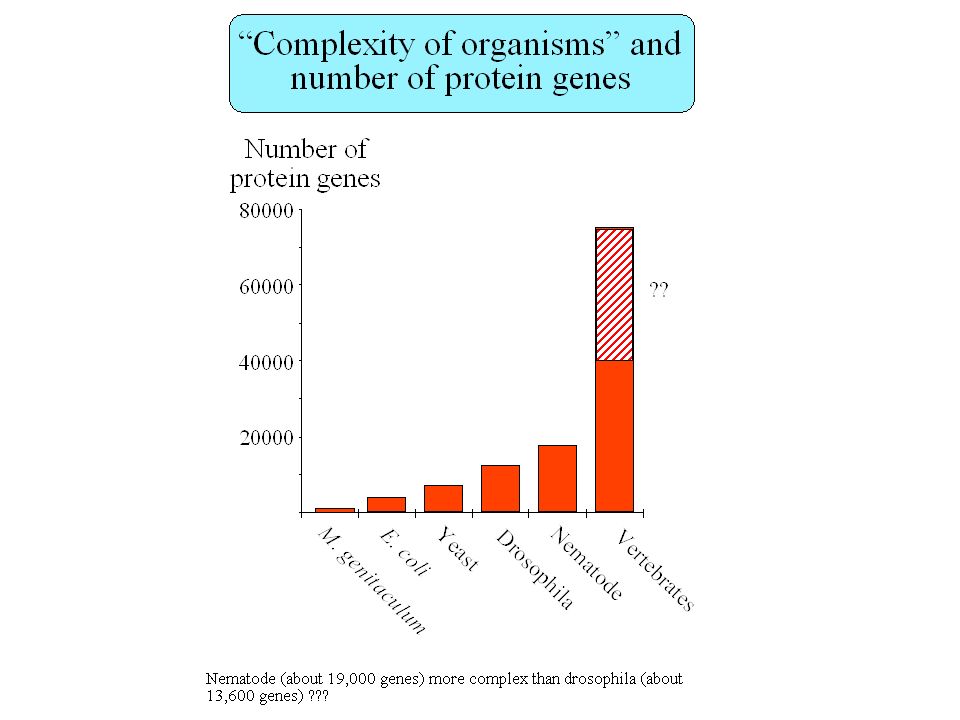
How many genes in the human genome ? Fields et al. 1994
Liang et al. (2000) 120,000 EST Erwing & Green (2000) 34,000 EST + chrom. 22 Roest Crollius et al. (2000) 31,000 Fish / human
 120,000 EST. Erwing & Green (2000) 34,000 EST + chrom. 22. Roest Crollius et al. (2000) 31,000 Fish / human.")
9
Structure of human protein genes
1396 complete human genes (exons + introns) from GenBank (1999) Average size (25%, 75%) Gene 15 kb ± 23 kb (4, 16) (10% > 35 kb) CDS nt ± 1200 (600, 1500) Exon (coding) 200 nt ± 180 (110, 200) Intron nt ± 3000 (500, 2000) 5'UTR 210 nt (Pesole et al. 1999) 3'UTR 740 nt (Pesole et al. 1999) Intron/exon Number of introns: 6 ±3 introns / kb CDS Introns / (introns + CDS): 80% 5' introns in 15% of genes (more ?), 3 ’introns very rare Alternative splicing in more than 30% of human genes (Hanke et al. 1999)
 from GenBank (1999) Average size (25%, 75%) Gene 15 kb ± 23 kb (4, 16) (10% > 35 kb) CDS 1300 nt ± 1200 (600, 1500) Exon (coding) 200 nt ± 180 (110, 200) Intron 1800 nt ± 3000 (500, 2000) 5 UTR 210 nt (Pesole et al. 1999) 3 UTR 740 nt (Pesole et al. 1999) Intron/exon. Number of introns: 6 ±3 introns / kb CDS. Introns / (introns + CDS): 80% 5 introns in 15% of genes (more ), 3 ’introns very rare. Alternative splicing in more than 30% of human genes (Hanke et al. 1999)")
10
Structure of human protein genes
GenBank: bias towards short genes 1396 complete human genes (exons + introns)
")
11
Structure of human protein genes
GenBank: bias towards short genes 1396 complete human genes (exons + introns) 9268 complete human mRNA
 9268 complete human mRNA.")
14
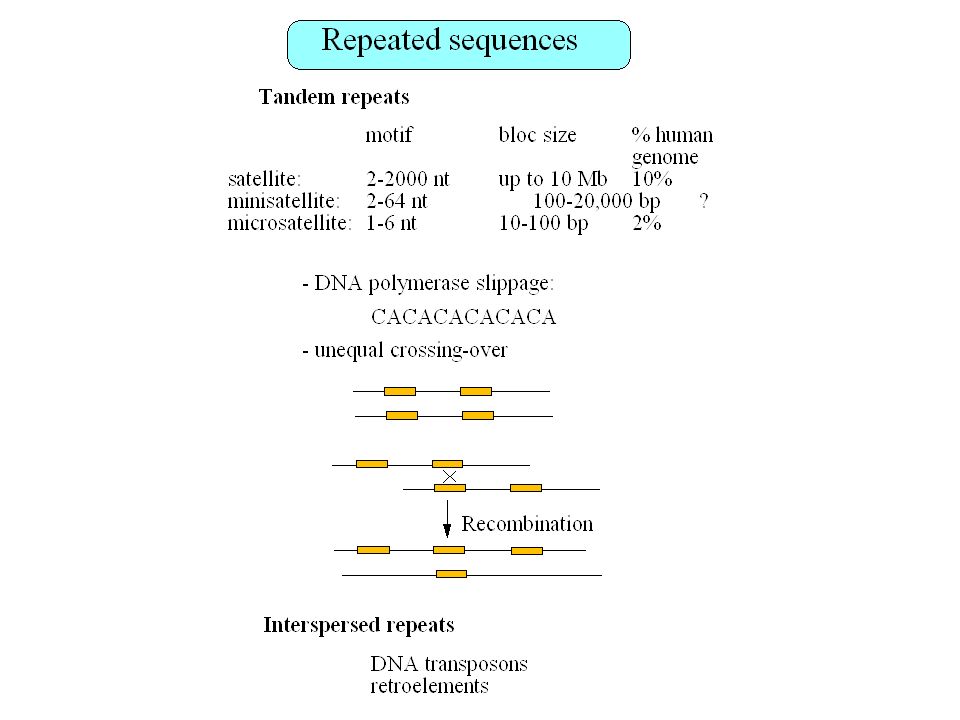
ADN satellite: centromères

18
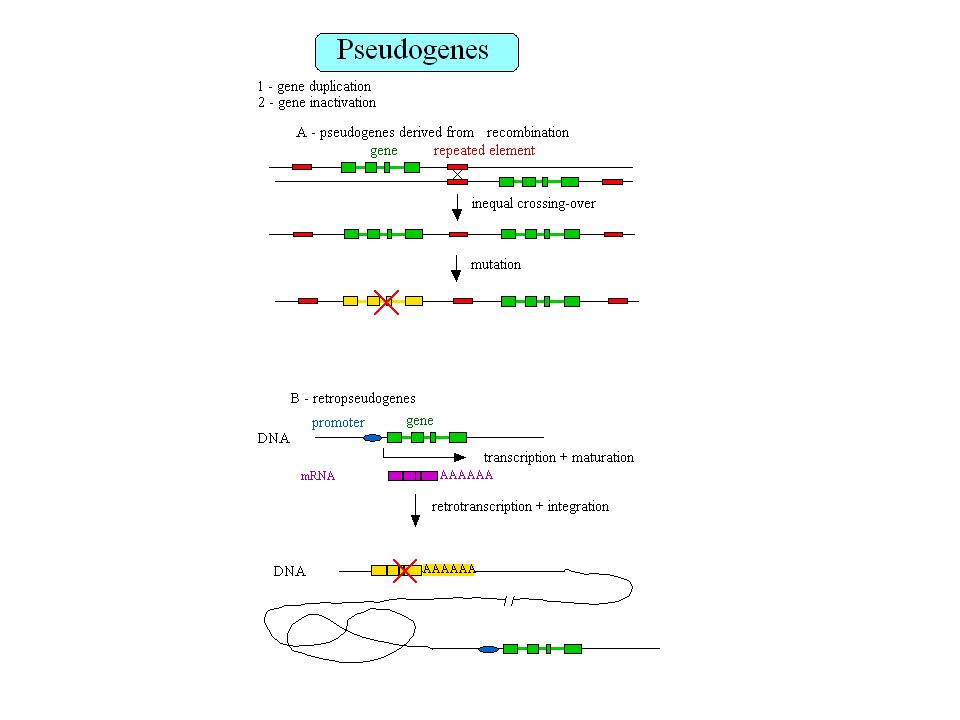
Retropseudogènes 23,000 à 33,000 retropseudogènes dans le génome humain Les gènes qui génèrent des retropseudogènes sont généralement de type housekeeping Gonçalves et al. 2000

19
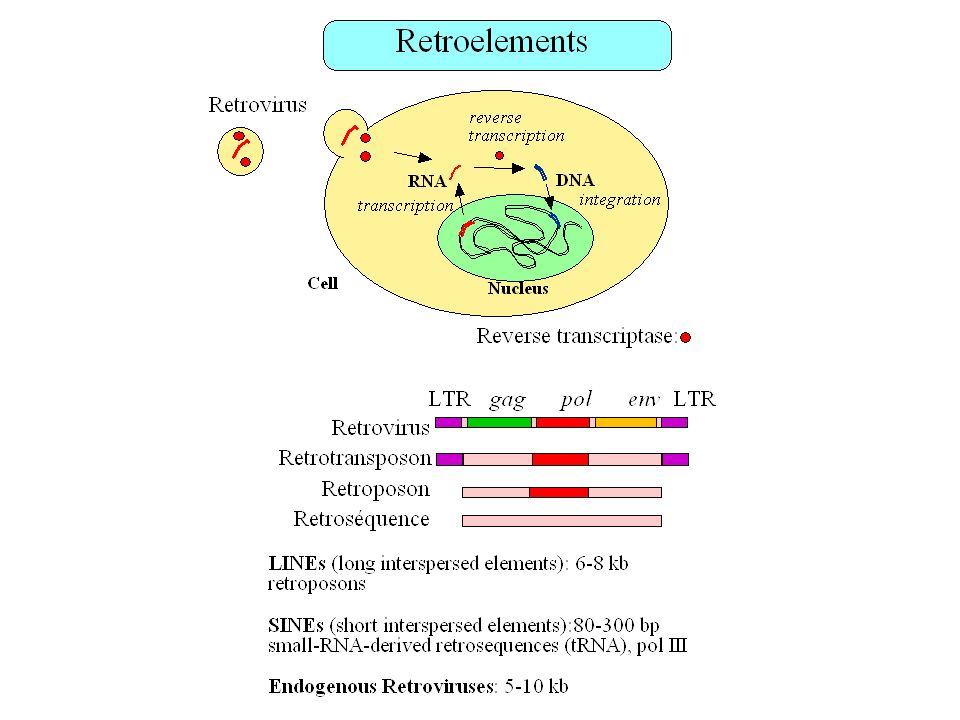
Fréquence des éléments transposables dans le génome humain
Total = 42% (Smit 1999)
")
20
Fréquence des éléments transposables dans le génome humain (Smit 1999)
")
21
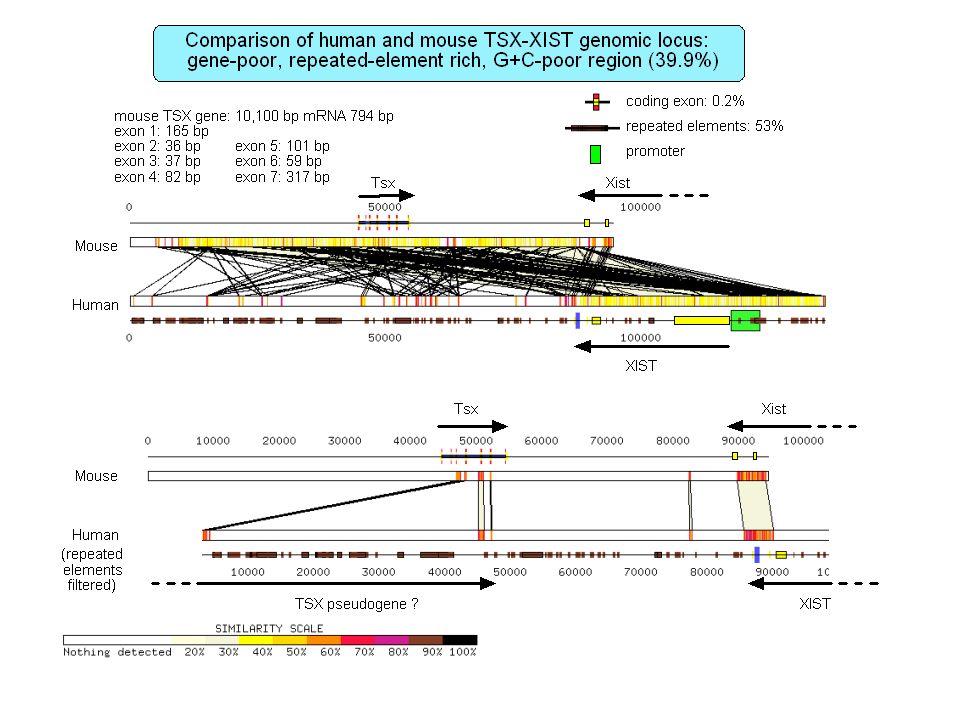
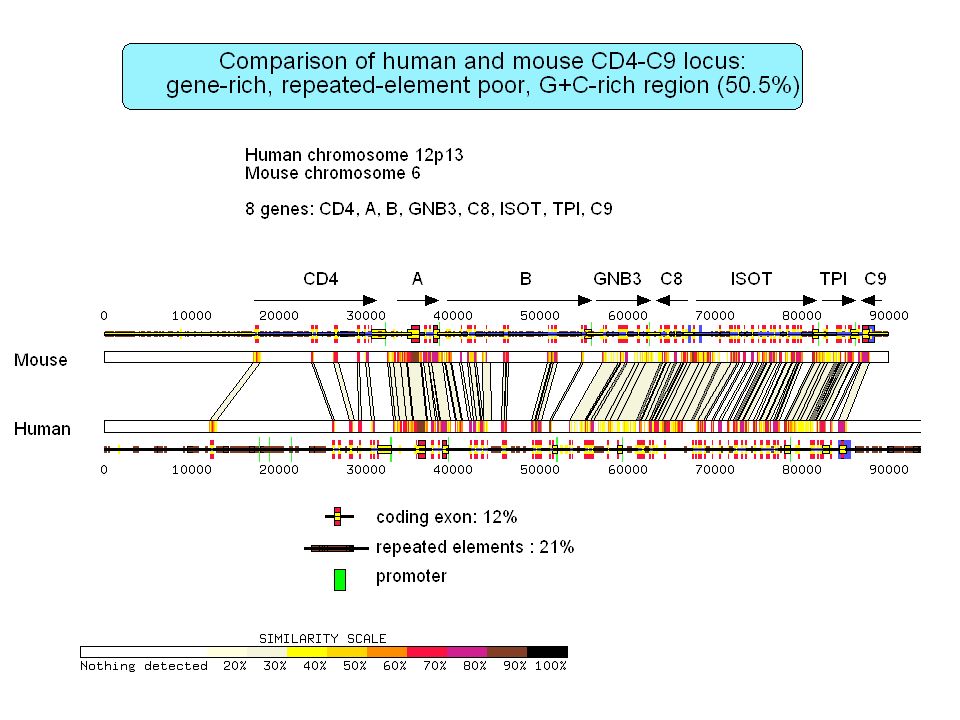
Isochore organization of vertebrate genomes

22
Organisation en isochore des génomes de vertébrés: mise en évidence expérimentale
Fractionnement du génome de la souris par centrifugation en gradient de densité (Bernardi et al. 1976)
")
23
Analyse statistique des séquences publiées dans les banques de données
Analyse statistique des séquences publiées dans les banques de données. Corrélation entre la composition en base en position 3 des codons et celle de l'envirronement génomique dans lequel se trouve le gène

24
Analyse statistique des séquences publiées dans les banques de données
Analyse statistique des séquences publiées dans les banques de données. Distribution en fréquence des gènes dans les différentes classes d'isochores 14 12 Moy = .580 Moy = .509 12 Ecart-t = .106 Ecart-t = .103 10 703 séq 10 173 séq 8 8 6 6 4 4 2 2 20 40 60 80 100 20 40 60 80 100 Nb de gènes (%) Danio Xénope 1 2 3 4 5 6 7 20 40 60 80 100 Moy = .612 Ecart-t = .158 5447 séq Moy = .639 Ecart-t = .171 818 séq Homme Poulet CDS GC3%
 Danio. Xénope Moy = Ecart-t = séq. Moy = Ecart-t = séq. Homme. Poulet. CDS GC3%")
25
Evolution de la structure en isochore chez les vertébrés

26
Isochore organization of vertebrate genomes
Insertion of repeated sequences (A. Smit 1996) Recombination frequency (Eyre-Walker 1993) Chromosome banding (Saccone, 1993) Replication timing (Bernardi, 1998) Gene density (Mouchiroud, 1991) Gene expression ?? -> No Gene structure (Duret, 1995)
 Recombination frequency (Eyre-Walker 1993) Chromosome banding (Saccone, 1993) Replication timing (Bernardi, 1998) Gene density (Mouchiroud, 1991) Gene expression -> No. Gene structure (Duret, 1995)")
27
Isochores and insertion of repeat sequences (Smit 1999)
4419 human genomic sequences > 50 kb

28
Isochores and gene density
MHC locus (3.6 Mb) (The MHC sequencing consortium 1999) Class I, class II (H1-H2 isochores): 20 genes/Mb, many pseudogenes Class III (H3 isochore): 84 genes/Mb, no pseudogene Class II boundaries correlate with switching of replication timing Chromosome 21
 (The MHC sequencing consortium 1999) Class I, class II (H1-H2 isochores): 20 genes/Mb, many pseudogenes. Class III (H3 isochore): 84 genes/Mb, no pseudogene. Class II boundaries correlate with switching of replication timing. Chromosome 21.")
29
Isochores and introns length
Duret, Mouchiroud and Gautier, 1995 760 complete human genes L1L2: intron G+C content < 46% H1H2: intron G+C content 46-54% H3: intron G+C content >54%

30
Projets Génome: état des lieux
Les différents projets génome Nature et qualité des séquences produites Annotation des séquences génomiques: recherche de régions fonctionnelles

32
Sequencing Projects : Genome / Transcriptome

33
Projets Génome Eubacteria: 29 génomes complets (dont 19 dans les 12 derniers mois !) Archaea: 6 génomes complets Eukaryotes: 3 (4) génomes complets levure: Mb 100% P. falciparum Mb 24% C. elegans (nematode) 100 Mb 95% A. thaliana Mb 60% Drosophila Mb 60% (100%) homme Mb 25% « draft » 90% complete in 2000, finished in 2003 souris Mb 1%
 génomes complets. levure: 13 Mb 100% P. falciparum 30 Mb 24% C. elegans (nematode) 100 Mb 95% A. thaliana 120 Mb 60% Drosophila 170 Mb 60% (100%) homme 3200 Mb 25% « draft » 90% complete in 2000, finished in souris 3000 Mb 1%")
34
Stratégie de séquençage du génome humain (projet public)
")
35
Etapes du séquençage génomique
ébauche (draft) Phase 0-1: séquence non-terminée; contigs non-ordonnés, non-orientés; gaps Phase 2: séquence non-terminée; contigs ordonnés, orientés; gaps Phase 3: séquence terminée Phase 0-2: séquences mentionnées HTG (High Throughput Genome sequences) dans les banques de données
 Phase 0-1: séquence non-terminée; contigs non-ordonnés, non-orientés; gaps. Phase 2: séquence non-terminée; contigs ordonnés, orientés; gaps. Phase 3: séquence terminée. Phase 0-2: séquences mentionnées HTG (High Throughput Genome sequences) dans les banques de données.")
36
Projets EST (transcriptome)
Expressed Sequence Tags (EST) Inventaire des ARNm exprimés par un organisme, dans différents tissus, stades de développement, pathologies, … Extraction et clonage des ARNm (banques d ’ADNc) Séquençage systématique des clones Séquences partielles d ’ARNm ( nt) Erreurs de séquence (1-3%) Redondance (gènes fortement exprimés) Qualité suffisante pour identifier un gène Automatisation
 Inventaire des ARNm exprimés par un organisme, dans différents tissus, stades de développement, pathologies, … Extraction et clonage des ARNm (banques d ’ADNc) Séquençage systématique des clones. Séquences partielles d ’ARNm ( nt) Erreurs de séquence (1-3%) Redondance (gènes fortement exprimés) Qualité suffisante pour identifier un gène. Automatisation.")
37
Projets EST à grande échelle
Nombre d'EST (Nov. 99) Homo sapiens ,611,810 Mus musculus (souris) ,486 Rattus sp. (rat) ,378 Caenorhabditis elegans (nématode) ,232 Drosophila melanogaster ,121 Oryza sativa (riz) ,083 Arabidopsis thaliana (arabette) ,752 Danio rerio (poisson zèbre) ,001 Zea mays (maïs) ,285 Lycopersicon esculentum (tomate) ,047
 Homo sapiens 1,611,810. Mus musculus (souris) 697,486. Rattus sp. (rat) 124,378. Caenorhabditis elegans (nématode) 101,232. Drosophila melanogaster 86,121. Oryza sativa (riz) 47,083. Arabidopsis thaliana (arabette) 45,752. Danio rerio (poisson zèbre) 40,001. Zea mays (maïs) 39,285. Lycopersicon esculentum (tomate) 38,047.")
38
Projets GSS Genome Survey Sequence (GSS)
Echantillonage aléatoire de séquence génomiques: donner un premier aperçu du contenu d'un génome Banques d ’ADN génomique Séquençage systématique de clones Séquences courtes (< 1kb) Erreurs de séquence (1-3%) Qualité suffisante pour identifier un gène Automatisation
 Erreurs de séquence (1-3%) Qualité suffisante pour identifier un gène. Automatisation.")
39
Projets GSS à grande échelle
Nombre de GSS (Nov. 2000) Homo sapiens ,041 Mus musculus (souris) ,033 Tetraodon nigroviridis ,963 Oryza sativa (riz) ,107 Strongylocentrotus purpuratus (oursin) ,019 Arabidopsis thaliana (arabette) ,265 Trypanosoma brucei ,123 Drosophila melanogaster ,785 Takifugu rubripes ,929
 Homo sapiens 863,041. Mus musculus (souris) 452,033. Tetraodon nigroviridis 188,963. Oryza sativa (riz) 93,107. Strongylocentrotus purpuratus (oursin) 76,019. Arabidopsis thaliana (arabette) 61,265. Trypanosoma brucei 48,123. Drosophila melanogaster 44,785. Takifugu rubripes 42,929.")
40
Genome Sequence Data Traditional sequences: correspond to biologically characterized genes, annotated by reearchers or database curators, usually relatively short (<20,000). Finished genome sequences: long contiguous sequences, correspond to clones (cosmid, BAC, PAC); partly automatically generated annotations covers repetitive elements, kown and predicted genes, EST matches Unfinished genome sequences (draft): large sequence entries consisting of unordered pieces separated by runs of N's, correspond to clones, contain minimal annotation. Genome survey sequences: low-quality, single pass sequences from a variety of different projects (BAC end sequencing, polymorphism studies, CpG islands, etc.), minimal annotation.
. Finished genome sequences: long contiguous sequences, correspond to clones (cosmid, BAC, PAC); partly automatically generated annotations covers repetitive elements, kown and predicted genes, EST matches. Unfinished genome sequences (draft): large sequence entries consisting of unordered pieces separated by runs of N s, correspond to clones, contain minimal annotation. Genome survey sequences: low-quality, single pass sequences from a variety of different projects (BAC end sequencing, polymorphism studies, CpG islands, etc.), minimal annotation.")
41
Different types of nucleotide sequences in current databases

42
GenBank release 119 (September 28, 2000)
Division Entries Nucleotides % nt EST ,843, ,337,244, % HTG , ,373,497, % GSS ,724, ,450, % PRI , ,073,472, % Other , ,296,473, % Total ,664, ,032,139, % Human ,518, ,253,704, %

43
The human genome sequencing project Where are we today (July 17 2000) ?
According to Phillip Bucher (SIB, Lausanne) statistics and genome coverage estimates (see also EBI's statistics: genome-MOT)
 statistics and genome coverage estimates (see also EBI s statistics: genome-MOT)")
44
Exponential growth of sequence data
Doubling time: 13 mounths (8 mounths) Publicly available sequences (Mb)
 Publicly available sequences (Mb)")
45
Contenu des banques de séquences nucléiques
Avril 2000: 6.106 séquences 7.109 bases espèces 9 espèces (0.02%) représentent à elles seules 85% des séquences Homo sapiens % Mus musculus % Drosophila melanogaster % Caenorhabditis elegans % Arabidopsis thaliana % Oryza sativa % Rattus norvegicus % Danio rerio % Saccharomyces cerevisiae %
 représentent à elles seules 85% des séquences. Homo sapiens 62.1% Mus musculus 7.7% Drosophila melanogaster 6.1% Caenorhabditis elegans 3.3% Arabidopsis thaliana 2.9% Oryza sativa 1.3% Rattus norvegicus 0.8% Danio rerio 0.6% Saccharomyces cerevisiae 0.6%")
46
Next steps in genome projects
Identify genes and other functional elements within genomic sequence (where are the genes ?) Determine the function of genes (what do they do ?)
 Determine the function of genes (what do they do )")
47
Structure of human protein genes
1396 complete human genes (exons + introns) from GenBank Average size (25%, 75%) Gene 15 kb ± 23 kb (4, 16) (10% > 35 kb) CDS nt ± 1200 (600, 1500) Exon (coding) 200 nt ± 180 (110, 200) Intron nt ± 3000 (500, 2000) 5'UTR 210 nt (Pesole et al. 1999) 3'UTR 740 nt (Pesole et al. 1999) Intron/exon Number of introns: 6 ±3 introns / kb CDS Introns / (introns + CDS): 80% 5' introns in 15% of genes (more ?), 3 ’introns very rare Alternative splicing in more than 30% of human genes (Hanke et al. 1999)
 from GenBank. Average size (25%, 75%) Gene 15 kb ± 23 kb (4, 16) (10% > 35 kb) CDS 1300 nt ± 1200 (600, 1500) Exon (coding) 200 nt ± 180 (110, 200) Intron 1800 nt ± 3000 (500, 2000) 5 UTR 210 nt (Pesole et al. 1999) 3 UTR 740 nt (Pesole et al. 1999) Intron/exon. Number of introns: 6 ±3 introns / kb CDS. Introns / (introns + CDS): 80% 5 introns in 15% of genes (more ), 3 ’introns very rare. Alternative splicing in more than 30% of human genes (Hanke et al. 1999)")
48
Prédiction de gènes eucaryotes (1) méthodes ab initio
Prédiction d ’exons codants Recherche de phases ouvertes de lecture (ORF: open reading frame) Taille moyenne des exons: ± 150 nt Statistiques sur les nucléotides, usage des codons Périodicité d'ordre 3, fréquence d ’hexamères Signaux d ’épissage Construction d ’un modèle de gène protéique Combinaison d ’exons de phases compatibles (pondération en fonction des scores de chaque exon potentiel) Recherche de limites de gènes Exons terminaux (5 ’, 3 ’) Promoteur Signal de polyadénylation Epissage alternatif ?? Exons non codants ?? Gène transcrits non codants (Xist, …) ??
 Taille moyenne des exons: ± 150 nt. Statistiques sur les nucléotides, usage des codons. Périodicité d ordre 3, fréquence d ’hexamères. Signaux d ’épissage. Construction d ’un modèle de gène protéique. Combinaison d ’exons de phases compatibles (pondération en fonction des scores de chaque exon potentiel) Recherche de limites de gènes. Exons terminaux (5 ’, 3 ’) Promoteur. Signal de polyadénylation. Epissage alternatif Exons non codants Gène transcrits non codants (Xist, …)")
49
Prédiction de gènes eucaryotes: qualité de la prédiction
Comparaison des différents logiciels: sensibilité/spécificité Sn: sensibilité Sp: spécificité par exon (sn_e, sp_e) ou par nucéotide (sn_e, sp_e) Jeu de données Burset-Guigo (1996): 570 gènes de vertébrés Jeu de données Salamov et al (1998): 660 gènes humains
 ou par nucéotide (sn_e, sp_e) Jeu de données Burset-Guigo (1996): 570 gènes de vertébrés. Jeu de données Salamov et al (1998): 660 gènes humains.")
50
Prédiction de gènes eucaryotes: qualité de la prédiction
Comparaison des différents logiciels: sensibilité/spécificité Sn: sensibilité Sp: spécificité par exon (sn_e, sp_e) ou par nucéotide (sn_e, sp_e) Locus BRCA2 (1.4 Mb, chrom. 13q) (Sanger Centre 1999): région "difficile" pour les logiciels de prédiction. 159 exons
 ou par nucéotide (sn_e, sp_e) Locus BRCA2 (1.4 Mb, chrom. 13q) (Sanger Centre 1999): région difficile pour les logiciels de prédiction. 159 exons.")
51
Prédiction de gènes protéiques complets
C. elegans: la plupart des ‘ gènes ’ annotés sont seulement des prédictions: 50% de prédictions fausses (dont la moitié sont exploitables) (J. Thierry-Mieg) Peut-on utiliser ces méthodes pour annoter les séquences génomique humaines ? + les faux positifs ! + épissage alternatif
 (J. Thierry-Mieg) Peut-on utiliser ces méthodes pour annoter les séquences génomique humaines + les faux positifs ! + épissage alternatif.")
52
Un peu d ’optimisme Fraction de la longueur des gènes correctement prédits: 70-80% Probabilité que deux exons potentiels consécutifs soient réels (et donc positifs en RT-PCR) 0.5
 0.5.")
53
Prediction of functional elements (2): ESTs
Large scale transcriptome projects: ESTs, full-length cDNA Identification of transcribed genes (protein or non-coding RNA) Information on alternative splicing, polyadenylation (Hanke et al. 1999, Gautheret et al. 1998), expression pattern SIM4: align a cDNA to genomic DNA Very useful but ... Problems with genes expressed at low level, narrow tissue distribution, stage-specific expression, … Limited tissue sampling Artifacts in ESTs (introns, partially matured RNA, …) Limited to polyadenylated RNA
 Information on alternative splicing, polyadenylation (Hanke et al. 1999, Gautheret et al. 1998), expression pattern. SIM4: align a cDNA to genomic DNA. Very useful but ... Problems with genes expressed at low level, narrow tissue distribution, stage-specific expression, … Limited tissue sampling. Artifacts in ESTs (introns, partially matured RNA, …) Limited to polyadenylated RNA.")
54
Prédiction de gènes eucaryotes (suite): approche comparative
tirer des informations de l'histoire évolutive des gènes 3,5 milliard d'années d'évolution MUTATIONS: - substitutions - insertions ou délétions locales - recombinaisons intergéniques, brassage d'exons - duplications ou pertes de gènes - pertes ou duplications de chromosomes - duplications de génomes - transgénèse (transfert horizontal de gènes) - etc.
 - etc.")
55
dérive génétique EVOLUTION MOLECULAIRE
"Nothing happens, but if it does it's not important" Dan Graur - les individus qui portent des mutations délétères n'ont pas ou peu de descendance. Ces mutations disparaissent rapidement de la population: sélection - les individus qui portent des mutations non (ou faiblement) délétère peuvent transmettre ces mutations à leur descendance. Ces mutations, dites "neutres", sont fixées aléatoirement dans la population (i.e. la majorité des individus portent ce génotype mutant): dérive génétique - les très rares mutations avantageuses sont positivement sélectionnées Les génomes sont le fruit de ces deux forces évolutives: sélection dérive génétique
 délétère peuvent transmettre ces mutations à leur descendance. Ces mutations, dites neutres , sont fixées aléatoirement dans la population (i.e. la majorité des individus portent ce génotype mutant): dérive génétique. - les très rares mutations avantageuses sont positivement sélectionnées. Les génomes sont le fruit de ces deux forces évolutives: sélection. dérive génétique.")
56
Prédiction des régions fonctionnelles (gènes protéiques, ARN, régions régulatrices) (2)
Comparative sequence analysis (phylogenetic footprinting) Function => selective pressure Corollary Sequence conservation = selective pressure = function provided the number of aligned homologous sequences represents enough evolutionary time for the accumulation of mutations at the less constrained (presumably selectively neutral) base positions. Evolutionary rate in non-functional DNA: ~ 0.3% / My (± 0.069) Man/Mouse: ~ 80 Myrs 46-58% identity Mammals/Birds: ~ 300 Myr 26-28% identity Random sequences 25% identity
 Function => selective pressure. Corollary. Sequence conservation = selective pressure = function. provided the number of aligned homologous sequences represents enough evolutionary time for the accumulation of mutations at the less constrained (presumably selectively neutral) base positions. Evolutionary rate in non-functional DNA: ~ 0.3% / My (± 0.069) Man/Mouse: ~ 80 Myrs 46-58% identity. Mammals/Birds: ~ 300 Myr 26-28% identity. Random sequences 25% identity.")
57
Prédiction de gènes eucaryotes (3)
Approche comparative Comparaison d ’une séquence génomique avec des gènes déjà caractérisés dans d ’autres espèces BLASTX WISE2: alignement ADN/protéine avec épissage) Comparaison de séquences génomiques (non-annotées) homologues Locus mnd2 (homme souris) (Jang et al. 1999): >80 kb Prédiction d ’exons internes basée sur la conservation de séquence ORF ≥ 80 nt Séquence protéique ≥ 70% similarité Séquence ADN ≥50% identité GT AG conservés => détection de tous les exons internes du gène D6Mm5e Généralisation de la méthode: ROSETTA (Genome Res :950) ALFRESCO (Genome Res :1148) WABA (Genome Res :1115)
 Comparaison de séquences génomiques (non-annotées) homologues. Locus mnd2 (homme souris) (Jang et al. 1999): >80 kb. Prédiction d ’exons internes basée sur la conservation de séquence. ORF ≥ 80 nt. Séquence protéique ≥ 70% similarité. Séquence ADN ≥50% identité. GT AG conservés. => détection de tous les exons internes du gène D6Mm5e. Généralisation de la méthode: ROSETTA (Genome Res :950) ALFRESCO (Genome Res :1148) WABA (Genome Res :1115)")
60
Prédiction de régions régulatrices
Méthodes ab initio Prédiction de promoteurs Îlots CpG Approche comparative

61
Prédiction de promoteurs eucaryotes
Combinaison de sites de fixation de facteur de transcription (ordre, orientation, distance) Motifs courts, dégénérés Difficile de distinguer les vrais sites des faux positifs: Motif à 4 bases: ≈1/256 pb (1/128 pb sur les deux brins) Boîtes TATA, CAAT , GC: absents dans beaucoup de promoteurs Banques de données de sites de fixation de facteurs de transcription (TRANSFAC), de promoteurs caractérisés expérimentalement (EPD) PromoterScan (Prestridge 1995): Mesure de la densité en sites potentiels de fixation de facteurs de transcription de long de la séquence (pondération en fonction de la fréquence des sites dans ou en dehors des vrais promoteurs)
 Motifs courts, dégénérés. Difficile de distinguer les vrais sites des faux positifs: Motif à 4 bases: ≈1/256 pb (1/128 pb sur les deux brins) Boîtes TATA, CAAT , GC: absents dans beaucoup de promoteurs. Banques de données de sites de fixation de facteurs de transcription (TRANSFAC), de promoteurs caractérisés expérimentalement (EPD) PromoterScan (Prestridge 1995): Mesure de la densité en sites potentiels de fixation de facteurs de transcription de long de la séquence (pondération en fonction de la fréquence des sites dans ou en dehors des vrais promoteurs)")
62
Prédiction de promoteurs: sensibilité, spécificité
Sensibilité: fraction des promoteurs qui sont trouvés par le logiciel PromoterScan: sensibilité = 70% (promoteurs à boîte TATA) Spécificité: fraction des vrais promoteurs parmi ceux qui ont été prédits PromoterScan: spécificité = 20% Un faux positif / 10 kb Génome humain: ≈ gènes, ≈1 promoteur/30 kb
 Spécificité: fraction des vrais promoteurs parmi ceux qui ont été prédits. PromoterScan: spécificité = 20% Un faux positif / 10 kb. Génome humain: ≈ gènes, ≈1 promoteur/30 kb.")
63
Prédiction de promoteurs eucaryotes: recherches en cours
Prise en compte de l'orientation relative et des distances entre sites de fixation de facteurs de transcription COMPEL (Kolchanov 1998): banque de données d'éléments composites FastM : recherche dans une séquence génomique d'une combinaison de deux sites de fixation de facteurs de transcription à une distance définie l'un de l'autre Recherche de corrélations entre sites PromoterInspector (Werner 2000) Sensibilité: 40% Spécificité: 45% Combinaison recherche ab initio / approche comparative: recherche de sites potentiels parmi les régions conservées
: banque de données d éléments composites. FastM : recherche dans une séquence génomique d une combinaison de deux sites de fixation de facteurs de transcription à une distance définie l un de l autre. Recherche de corrélations entre sites. PromoterInspector (Werner 2000) Sensibilité: 40% Spécificité: 45% Combinaison recherche ab initio / approche comparative: recherche de sites potentiels parmi les régions conservées.")
64
Îlots CpG ou Génome de vertébrés : Me-C fortement mutable -> T
méthylation des C dans les dinucléotides 5 ’-CG-3 ’(CpG) Me-C fortement mutable -> T 5 ’-CG- 3 ’ 5 ’-TG-3 ’ ’-CA-3 ’ 3 ’-GC- 5 ’ ’-AC-5 ’ ’-GT-5 ’ Génome des vertébrés: globalement dépourvu en CpG (excès de TG, CA) Certaines régions (200 nt à plusieurs kb) échappent à la méthylation Pas de déplétion en CpG: CpGo/e proche de 1 Riche en G+C Îlot CpG: Longueur > 500 nt CpGo/e > 0.6 G+C > 50% ou
 Me-C fortement mutable -> T. 5 ’-CG- 3 ’ 5 ’-TG-3 ’ 5 ’-CA-3 ’ 3 ’-GC- 5 ’ 3 ’-AC-5 ’ 3 ’-GT-5 ’ Génome des vertébrés: globalement dépourvu en CpG (excès de TG, CA) Certaines régions (200 nt à plusieurs kb) échappent à la méthylation. Pas de déplétion en CpG: CpGo/e proche de 1. Riche en G+C. Îlot CpG: Longueur > 500 nt. CpGo/e > 0.6. G+C > 50% ou.")
65
La déamination des cytosines
CH H NH 2 O Uracile HN C CH N H O déamination réparation Cytosine N C CH H NH 2 O Cytosine méthylée N C CH H NH 2 O CH3 Thymine HN C CH N H O CH3 déamination TpG ou CpA

66
Îlots CpG: associé aux régions promotrices ?
Bird (1986), Gardiner-Garden (1987) Larsen (1992) ref 40% des gènes tissu-spécifiques possèdent un îlot CpG en 5 ’ 100% des gènes ‘ housekeeping ’ possèdent un îlot CpG en 5 ’ Rechercher des îlots CpG pour prédire des régions promotrices ? Sensibilité: % Spécificité ?? (Quelle fraction des îlots CpG correspond effectivement à des régions promotrices ?) Ioshikhes & Zhang (2000): comparaison des îlots CpG qui recouvrent ou non le site d ’initiation de la transcription
, Gardiner-Garden (1987) Larsen (1992) ref. 40% des gènes tissu-spécifiques possèdent un îlot CpG en 5 ’ 100% des gènes ‘ housekeeping ’ possèdent un îlot CpG en 5 ’ Rechercher des îlots CpG pour prédire des régions promotrices Sensibilité: % Spécificité (Quelle fraction des îlots CpG correspond effectivement à des régions promotrices ) Ioshikhes & Zhang (2000): comparaison des îlots CpG qui recouvrent ou non le site d ’initiation de la transcription.")
67
Fréquence des gènes humains avec un îlot CpG recouvrant le site d ’initiation de la transcription
800 gènes humains avec promoteur décrit Mesure de la distribution tissulaire à l ’aide d ’EST (20 tissus)
")
68
Comparaison des îlots CpG recouvrant ou non le site d ’initiation de la transcription
272 îlots start CpG recouvrant le site d ’initiation de la transcription (start) 1078 îlots CpG en dehors d ’un promoteur connu (other) (en excluant les séquences répétées)
 1078 îlots CpG en dehors d ’un promoteur connu (other) (en excluant les séquences répétées)")
69
Prediction of functional elements (2)
Comparative sequence analysis (phylogenetic footprinting) Function => selective pressure Corollary Sequence conservation = selective pressure = function provided the number of aligned homologous sequences represents enough evolutionary time for the accumulation of mutations at the less constrained (presumably selectively neutral) base positions. Evolutionary rate in non-functional DNA: ~ 0.3% / My (± 0.069) Man/Mouse: ~ 80 Myrs 46-58% identity Mammals/Birds: ~ 300 Myr 26-28% identity Random sequences 25% identity
 Function => selective pressure. Corollary. Sequence conservation = selective pressure = function. provided the number of aligned homologous sequences represents enough evolutionary time for the accumulation of mutations at the less constrained (presumably selectively neutral) base positions. Evolutionary rate in non-functional DNA: ~ 0.3% / My (± 0.069) Man/Mouse: ~ 80 Myrs 46-58% identity. Mammals/Birds: ~ 300 Myr 26-28% identity. Random sequences 25% identity.")
70
Analyse comparative des gènes de b-actine de l'homme et de la carpe

71
Recherche de régions régulatrices par analyse comparative (empreintes phylogénétiques)
Goodman et al. 1988: régulation de l’expression des gènes du cluster b-globine au cours du développement Alignement de séquences orthologues de 6 mammifères (> 270 Ma d’évolution) 13 empreintes phylogénétiques: ≥ 6 nt, conservation 100% Analyse par retard de bande sur gel: 12/13 (92%) correspondent à des sites de fixation de protéines 1996: 35 empreintes phylogénétiques avec protéines fixatrices identifiées Enhancers de gènes HOX (Fugu/souris) (Aparicio et al. 1995) enhancer TCR a (homme/souris) (Luo, 1998) promoteur COX5B (11 primates) (Bachman, 1996) promoteur uPAR (homme/souris) (Soravia, 1995)
 13 empreintes phylogénétiques: ≥ 6 nt, conservation 100% Analyse par retard de bande sur gel: 12/13 (92%) correspondent à des sites de fixation de protéines. 1996: 35 empreintes phylogénétiques avec protéines fixatrices identifiées. Enhancers de gènes HOX (Fugu/souris) (Aparicio et al. 1995) enhancer TCR a (homme/souris) (Luo, 1998) promoteur COX5B (11 primates) (Bachman, 1996) promoteur uPAR (homme/souris) (Soravia, 1995)")
72
Large scale phylogenetic footprinting
Non-coding sequences : 325,247 sequences Mb everything except protein-coding regions and structural RNA genes (rRNA, tRNA, snRNA, scRNA) Introns, 5' and 3' untranslated regions, intergenic sequences Filtering of microsatellite repeats and cloning vectors: XBLAST Similarity search: BLASTN + LFASTA Vertebrates, insects, nematode
 Introns, 5 and 3 untranslated regions, intergenic sequences. Filtering of microsatellite repeats and cloning vectors: XBLAST. Similarity search: BLASTN + LFASTA. Vertebrates, insects, nematode.")
73
Metazoan Genome Projects

74
Sequence Similarities
1- Identification of new genes protein-genes, RNA-genes: intronic snoRNA genes 2- Retroviral elements, retrotransposons 3- Low complexity sequences: GC-rich, AT-rich, cryptic microsatellites 4- Artefacts: annotation errors, sample contamination (sponge insulin, ascidian RNA, chicken TGFB1) highly conserved regions (HCRs) - do not code for proteins - do not correspond to any known structural RNA
 highly conserved regions (HCRs) - do not code for proteins. - do not correspond to any known structural RNA.")
75
326 Highly Conserved Regions (HCRs)
• > 70% identity over 50 to 2000 nt after more than 300 Myrs • Unique sequences • Generally specific of only one gene • Longest HCR: 84% identity over 1930 nt after 300 Myrs 3’UTR deltaEF1 transcription factor • Oldest HCRs: 500 to 600 Myrs • No HCR between vertebrates and insects or nematode

76
Oldest HCRs

77
Conservation pattern in 3’UTRs

78
Distribution of HCRs within genes

79
HCRs and multigenic families

80
Function of 3’HCRs: mRNA stability, translation

81
Function of 3’HCRs: mRNA subcellular localization
Myosin heavy chain, c-myc, vimentin, b-actin

82
Comparaison des régions non-codantes de 77 gènes orthologues homme/souris (Jareborg et al. 1999)
Fraction des régions non-codantes conservées entre homme et souris

83
Phylogenetic footprinting
Advantages Works for all kinds of functional elements (transcribed or not, coding or not) as far as the information is in the primary sequence Does not require any a priori knowledge of the functional elements Limits Absence of evolutionary conservation does not mean absence of function No efficient method to detect unknown conserved secondary structure in RNA Function, but what function ? Depends on the sequencing status of other genomes Human, mouse, fugu, C. elegans, drosophila, yeast, A. thaliana Number of sequences to compare : > 200 Myrs of evolution Mammals/birds: 310 Myrs Human + mouse + bovine : 240 Myrs
 as far as the information is in the primary sequence. Does not require any a priori knowledge of the functional elements. Limits. Absence of evolutionary conservation does not mean absence of function. No efficient method to detect unknown conserved secondary structure in RNA. Function, but what function Depends on the sequencing status of other genomes. Human, mouse, fugu, C. elegans, drosophila, yeast, A. thaliana. Number of sequences to compare : > 200 Myrs of evolution. Mammals/birds: 310 Myrs. Human + mouse + bovine : 240 Myrs.")
Présentations similaires













.>")









