Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
La Maladie d’Alzheimer:
Neuropathologie

2
Les trois stades de lésions cérébrales
Stade enthorinal Stade hippocampique Stade néocortical Stade entorhinal: les lésions sont confinées à une seule couche de la région entorhinale Stade hippocampique: les lésions envahissent l’hippocampe donnant lieu à un « oubli à mesure » Stade néorcortical: les lésions se diffusent au cortex, entraînant l’apparition des troubles instrumentaux

3
Taille FH normale/AD Formation hippocampique normale

4
Dégénérescence neuronale: des stades 1 à 10.

5
Neuropathologie Générale: Que se passe-t-il?
Accumulation de protéïnes bêta-amyloïdes à l’extérieur de la mémbrane du neurone; Accumulation de protéïnes Tau aux niveaux des axones et des dendrites.

6
Comment se fait l’accumulation de protéïnes bêta-amyloïdes?
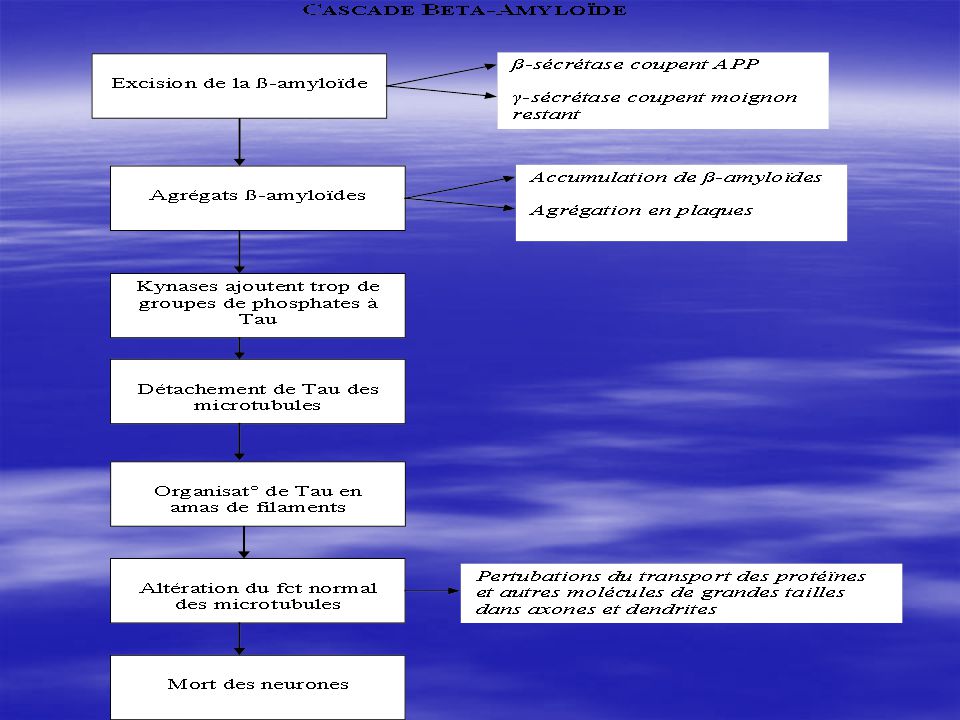
Les protéases bêta-secrétases coupent le précurseur de la protéïnes ß-amyloïdes (APP), à l’extérieur de la membrane, à l’aide de deux acides aspartiques. (fig) Les protéïnes présilinines, élt de la protéase gamma-sécrétases, coupent le moignon restant, à l’intérieur de la mémbrane, à l’aide de deux acides aspartiques, libérant ainsi la protéïne ß-amyloïde à l’ext. de la membrane. (fig) Les protéïnes ß-amyloïdes s’accumulent puis s’agrègent en plaques qui s’accumulent (fig).
, à l’extérieur de la membrane, à l’aide de deux acides aspartiques. (fig) Les protéïnes présilinines, élt de la protéase gamma-sécrétases, coupent le moignon restant, à l’intérieur de la mémbrane, à l’aide de deux acides aspartiques, libérant ainsi la protéïne ß-amyloïde à l’ext. de la membrane. (fig) Les protéïnes ß-amyloïdes s’accumulent puis s’agrègent en plaques qui s’accumulent (fig).")
7
Accumulation de la protéïne bêta-amyloïde
Les protéases bêta-secrétases coupent le précurseur de la protéïnes ß-amyloïdes (APP), à l’extérieur de la membrane, à l’aide de deux acides aspartiques Les protéïnes présilinines, élt de la protéase gamma-sécrétases, coupent le moignon restant, à l’intérieur de la mémbrane, à l’aide de deux acides aspartiques, libérant ainsi la protéïne ß-amyloïde à l’ext. de la membrane.
, à l’extérieur de la membrane, à l’aide de deux acides aspartiques. Les protéïnes présilinines, élt de la protéase gamma-sécrétases, coupent le moignon restant, à l’intérieur de la mémbrane, à l’aide de deux acides aspartiques, libérant ainsi la protéïne ß-amyloïde à l’ext. de la membrane.")
8
Création de filaments et de plaques

9
Comt se fait l’accumulation de protéïnes Tau?
L’accumulation de protéïnes ß-amyloïdes à l’ext. du neurone modifie l’activité chimique du neurone. En particulier, la kynase (une enzyme), ajoute trop de groupes de phospates la protéïne. Cette protéïne Tau modifiée se détache des microtubules et s’organisent en amas de filaments neurofibrillaires. Les protéïnes Tau, ainsi accumulées dans les microtubules, empêchent ces dernières d’accomplir leur f° normale qui est de transporter les protéïnes et d’autres molécules de grandes tailles à travers les axones et les dendrites. Ceci tue les neurones dès que proportion DNF trop importante.
, ajoute trop de groupes de phospates la protéïne. Cette protéïne Tau modifiée se détache des microtubules et s’organisent en amas de filaments neurofibrillaires. Les protéïnes Tau, ainsi accumulées dans les microtubules, empêchent ces dernières d’accomplir leur f° normale qui est de transporter les protéïnes et d’autres molécules de grandes tailles à travers les axones et les dendrites. Ceci tue les neurones dès que proportion DNF trop importante.")
10
Protéïne Tau: fonctionnement normal

11
Protéïnes Tau: Dysfonctionnement

13
Quelques faits à retenir dans la neuropathologie de la MA
Accumulation de plaques de protéïnes ß-amyloïdes Accumulation d’amas de filaments de protéïnes Tau Modifications neurochimiques: excision de l’APP par bêta- et gamma-sécrétases, ajout de trop de groupes de phosphates dans Tau par kynases Agrégation de protéïnes Tau en filaments Détachement des protéïnes Tau des microtubules Altérations de la f° normale des microtubules Mort des neurones

14
Voies thérapeutiques explorées
Inhibiteurs de protéases: molécules recherchées pour inhiber les protéases coupant la protéïne ß-amyloïde de son précurseur APP (mais ß-sécrétase joue rôle crucial dans différenciat° cellulaire) Anti-corps ß-amyloïde: introduction d’anti-corps dans le cerveau qui stimuleraient les cellules immunitaires du SNC (cellules de la microglie) à attaquer les aggrégats de la protéïne ß-amyloïde Blocage des enzymes qui fabriquent la protéïne ß-amyloïde Recherche de composés qui interagissent avec ß-amyloïde pour maintenir ce peptide à l’état dissous dans le liquide extracellulaire Etude de médicaments hypocholestérolémiants (statines) afin d’abaisser les qtités de cholestérol sanguin pour réduire la prod° de protéïnes précurseurs de ß-amyloïde et/ou pour inhiber l’activité des sécrétases. Thérapie cellulaire: Prélever peau, insérer dans ces cell. le gène codant le facteur de croissance nerveux (NGF). Placer ensuite ces cell. génétiqt modifiées dans le cerveau antérieur afin qu’elles y sécrètent du NGF et empêchent la perte de neurones (cholinergiques)
 Anti-corps ß-amyloïde: introduction d’anti-corps dans le cerveau qui stimuleraient les cellules immunitaires du SNC (cellules de la microglie) à attaquer les aggrégats de la protéïne ß-amyloïde. Blocage des enzymes qui fabriquent la protéïne ß-amyloïde. Recherche de composés qui interagissent avec ß-amyloïde pour maintenir ce peptide à l’état dissous dans le liquide extracellulaire. Etude de médicaments hypocholestérolémiants (statines) afin d’abaisser les qtités de cholestérol sanguin pour réduire la prod° de protéïnes précurseurs de ß-amyloïde et/ou pour inhiber l’activité des sécrétases. Thérapie cellulaire: Prélever peau, insérer dans ces cell. le gène codant le facteur de croissance nerveux (NGF). Placer ensuite ces cell. génétiqt modifiées dans le cerveau antérieur afin qu’elles y sécrètent du NGF et empêchent la perte de neurones (cholinergiques)")
15
Voies thérapeutiques?

16
Données génétiques Les porteurs d’un certain variant du gène codant l’apolipoprotéïne E (une protéïne qui favorise les assemblages de protéïnes ß-amyloïdes) ont un risque élevé de MA. Des mutations dans 2 gènes, Présilinine 1 et 2, sont responsables de formes précoces et fulgurantes de la MA. Les protéïnes codées par ces gènes sont intégrées dans l’enzyme γ-sécrétase. Chez les trisomique 21 (trois copies du chromos. 21), le chromos 21 contient le gène précurseur de la protéïne ß-amyloïde. C’est pourquoi les tris. 21 fabriquent plus de protéïnes ß-amyloïdes dès la naissance, ont des dépôts amyloïdes dès 12 ans et ont la MA à 50 ans.
 ont un risque élevé de MA. Des mutations dans 2 gènes, Présilinine 1 et 2, sont responsables de formes précoces et fulgurantes de la MA. Les protéïnes codées par ces gènes sont intégrées dans l’enzyme γ-sécrétase. Chez les trisomique 21 (trois copies du chromos. 21), le chromos 21 contient le gène précurseur de la protéïne ß-amyloïde. C’est pourquoi les tris. 21 fabriquent plus de protéïnes ß-amyloïdes dès la naissance, ont des dépôts amyloïdes dès 12 ans et ont la MA à 50 ans.")
17
Questions Quels sont les trois faits importants qui caractérisent la neuropathologie de la MA? Comment survient l’accumulation de plaques de protéïnes ß-amyloïdes? Quelles protéases coupent l’APP? Comment se fait l’accumulation de la protéïnes Tau? Quelle est la conséquence principale de l’accumulation de la protéïne Tau au niveau neurochimique? A quel moment le neurone meurt-il dans la MA?

Présentations similaires








>")










