Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
UNIVERSITE DE PICARDIE JULES VERNE Faculté de Pharmacie
Chimie Organique- EA3901-DMAG-INERIS J. Pecher, P. Sonnet, F.Y. Dupradeau MODELISATION MOLECULAIRE Mai 2005

2
A son balbutiement, la modélisation moléculaire était utilisée pour étudier :
La relations structure activité L’identification de pharmacophores Le design d’analogue La modélisation moléculaire intervient dans les différentes étapes de la chimie combinatoire

3
Trois types d’approches
Exploitation de la structure 3D de la cible biologique Structure 3D de la cible biologique inconnue Design de la librairie combinatoire

4
Le processus est multiparamétrique avec une optimisation simultanée des :
Propriétés biologiques (potentielle in vitro et in vivo) Propriétés physicochimiques (log P, pKa, liaison hydrogène, aire de surface, volume, réfractivité molaire polarisabilité molaire) Propriétés pharmaceutiques (solubilité, cristallinité) Propriétés pharmacocinétique (absorption, métabolisme, distribution et l’élimination)
 Propriétés physicochimiques (log P, pKa, liaison hydrogène, aire de surface, volume, réfractivité molaire polarisabilité molaire) Propriétés pharmaceutiques (solubilité, cristallinité) Propriétés pharmacocinétique (absorption, métabolisme, distribution et l’élimination)")
5
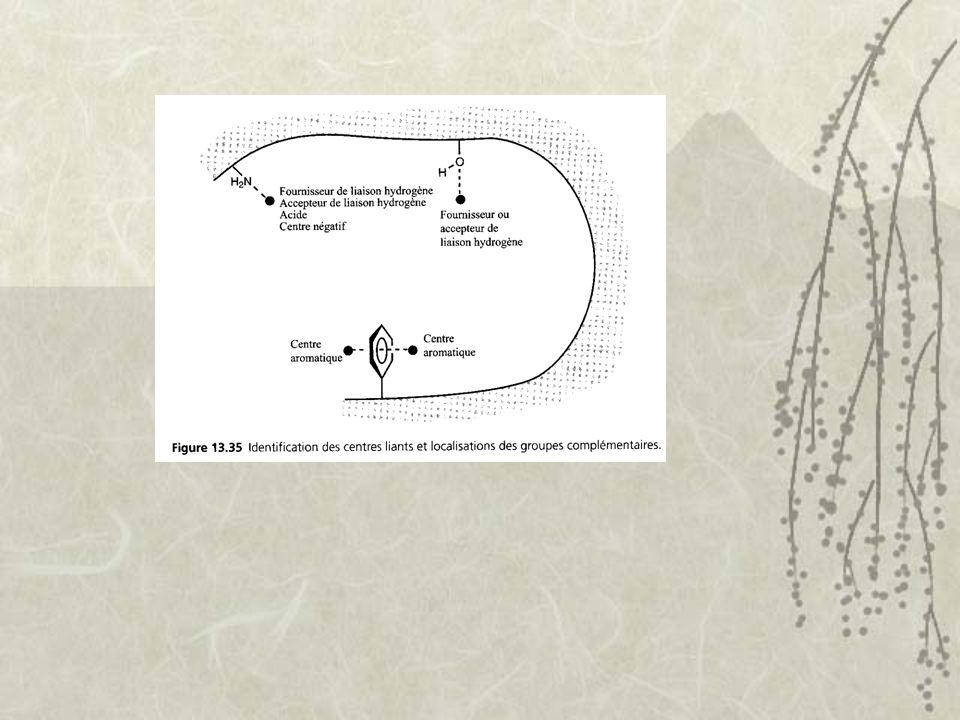
Pharmacophore : Fournisseur de liaison hydrogène (NH amide, amine aromatique et hydroxyle) Accepteur de liaison hydrogène (carbonyle etc.) Cycle aromatique Groupe acide (acide carboxylique, tetrazole non-substitué, acyl sulfonamide) Groupe basique (amine aliphatique, amidines/guanidines, 4-amino pyridine etc.) Région hydrophobe (isopropyle, butyle etc.)
 Groupe basique (amine aliphatique, amidines/guanidines, 4-amino pyridine etc.) Région hydrophobe (isopropyle, butyle etc.)")
6
Design de la bibliothèque combinatoire virtuelle
Similarité moléculaire Diversité moléculaire Rule of 5 ou la règle des 5 Utiliser des réactifs déterminer à partir de la similitude et la diversité moléculaire pour créer ainsi la bibliothèque combinatoire virtuelle

7
Structures générées similitude moléculaire
Donc structure très proche de la molécule originale Structures générées diversité moléculaire Donc structures plus eloignées de la molécule originale

8
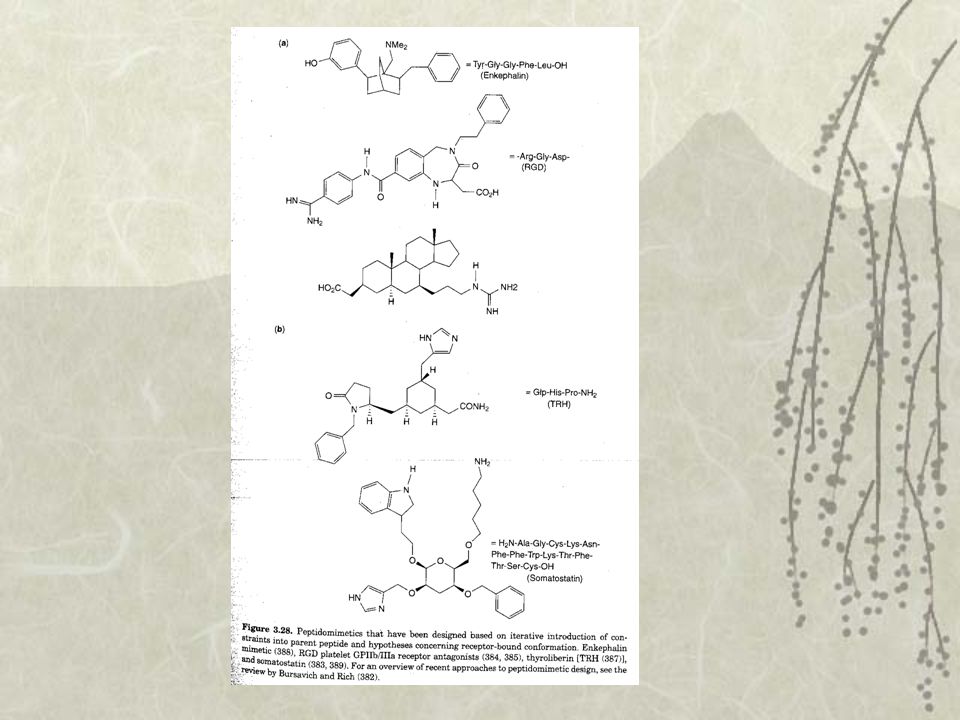
4 points du pharmacophore
Séquence RGD Diverses structures antagonistes du fibrinogène

9
La règle des 5 états est utilisé pour définir des prototypes idéaux
Absorption est plus probable quand il y a : 1) moins de 5 doneurs de liaisons hydrogènes 2) le poids moléculaire est inférieur à500 3) que le CLog P est inférieur à5 (ou MLOGP est inférieur de 4.15) 4) moins de 10 accepteur de liaisons hydrogènes (où les accepteurs sont les O et les N) Cette règle est ensuite utilisée pour construire les bibliothèques virtuelles En conclusion, la considération de la notation calculée P, le poids moléculaire, et la superficie polaire dans la conception des bibliothèques peuvent donner des composés avec des caractéristiques améliorées pour l’absorption et fournissent un outil valable au chimiste médicinal dans l'optimisation de la tête de série
 moins de 5 doneurs de liaisons hydrogènes. 2) le poids moléculaire est inférieur à500. 3) que le CLog P est inférieur à5 (ou MLOGP est inférieur de 4.15) 4) moins de 10 accepteur de liaisons hydrogènes (où les accepteurs sont les O et les N) Cette règle est ensuite utilisée pour construire les bibliothèques virtuelles. En conclusion, la considération de la notation calculée P, le poids moléculaire, et la superficie polaire dans la conception des bibliothèques peuvent donner des composés avec des caractéristiques améliorées pour l’absorption et fournissent un outil valable au chimiste médicinal dans l optimisation de la tête de série.")
10
Structure 3D de la cible biologique inconnue
Dynamique moléculaire pas possible Utilisation des données pharmacologiques Importance des groupements fonctionnelles par modification chimique et en testant leur activité biologique permet de déterminer des pharmacophores Peptidomimétique : amide remplacé par des sucres cycliques, stéroïdes, benzodiazépines ou bien encore des carbocycles Screening des récepteurs une fois que le pharmacophore est défini homologie de fixation avec une structure connue Déterminer une tête de série puis regarder la similitude et la diversité moléculaire

12
Exploitation de la structure 3D de la cible biologique
Connaître la structure de la protéine cible (RX, RMN) Caractérisation du site de fixation Design du ligand Calcul d’affinité
 Caractérisation du site de fixation. Design du ligand. Calcul d’affinité.")
13
Caractérisation du site de fixation
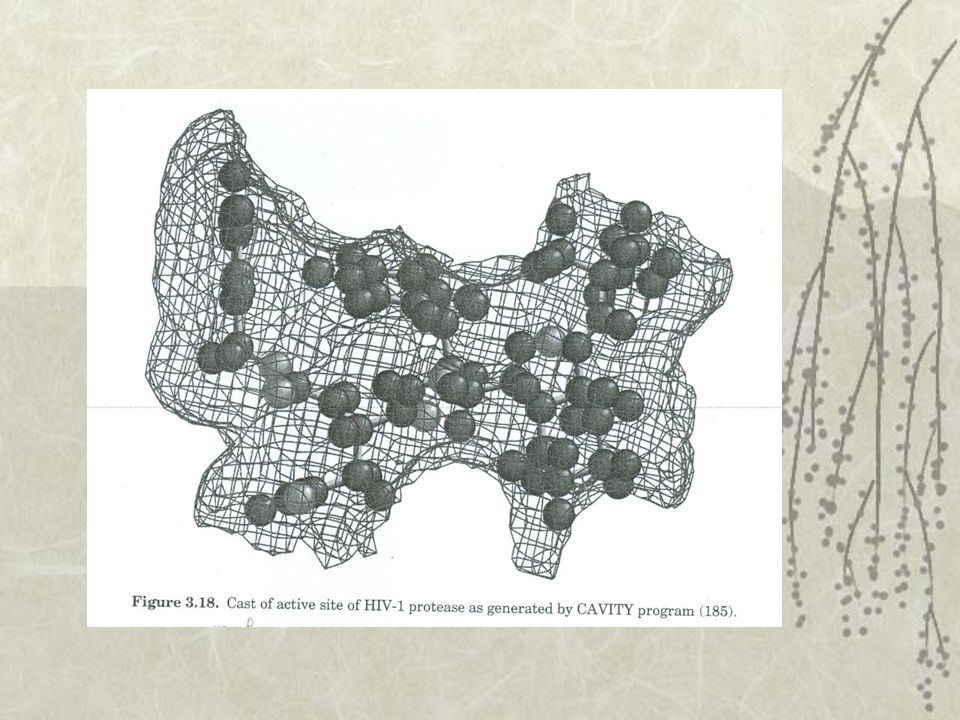
Volume et forme Déterminer la cavité, la poche enfouie dans la cible DOCK explore la complémentarité stérique entre un ligand connu et son récepteur Représentation schématique de la cavité

15
Potentiel électrostatique et hydrophobe
Liaison hydrogène GRID explore la cavité en estimant l’enthalpie d’interaction (donc permet de générer une carte du contour en 3D) CoMFA effectue une cartographie du site actif Permet de déterminer les liaisons hydrogènes possibles Potentiel électrostatique et hydrophobe CAVITY représente la surface d’hydrophobicité de la cavité et détermine la position où les interactions électrostatiques entre la cavité et le ligand
 CoMFA effectue une cartographie du site actif. Permet de déterminer les liaisons hydrogènes possibles. Potentiel électrostatique et hydrophobe. CAVITY représente la surface d’hydrophobicité de la cavité et détermine la position où les interactions électrostatiques entre la cavité et le ligand.")
16
Design du ligand Design assisté visuellement
Screenning des bases de données tridimensionnelles en utilisant CONCORD, MOLPAT, CAVEAT, ALADDIN, CHEM-X … Design de novo (basé sur la géométrie de composé cyclique comme core) et sur l’encombrement stérique : BRIDGE, CAVEAT, ALADDIN … Docking permet de déterminer les interactions possibles entre le ligand et le récepteur : DOCK
 et sur l’encombrement stérique : BRIDGE, CAVEAT, ALADDIN … Docking permet de déterminer les interactions possibles entre le ligand et le récepteur : DOCK.")
18
Exemple d’analogues du diphophoglycérate

19
Docking et Screening virtuelle d’un inhibiteur de la Proteinphosphatase 1B
Docking d’ inhibiteurs de l’acétylcholinestérase

Présentations similaires






>")





 n.>")




 des récepteurs NMDA (NMADRs) Paradigme du GluR2 pour laction.>")

