Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Anamnèse Madame N.A. âgée de 30 ans
Difficultés de décontraction musculaire (depuis l’âge de 13 ans) : mains, cuisses, paupières, langue « Muscles bloqués pendant qq secondes » Tous les 2 à 3 mois au début, quotidiennement aujourd’hui Détérioration très nette lors de la récente grossesse Difficultés à l’initiation d’un effort et après celui-ci (ex. Vélo) Le froid n’aggrave pas les symptômes Absence de paralysie (rares dérobements des M.Inf.) et de douleur
 : mains, cuisses, paupières, langue « Muscles bloqués pendant qq secondes » Tous les 2 à 3 mois au début, quotidiennement aujourd’hui. Détérioration très nette lors de la récente grossesse. Difficultés à l’initiation d’un effort et après celui-ci (ex. Vélo) Le froid n’aggrave pas les symptômes. Absence de paralysie (rares dérobements des M.Inf.) et de douleur.")
2
A.T.C.D. médico-chirurgicaux Toxiques et médicaments
Aucun A.T.C.D. héréditaires Père : 53 ans, lombalgies chroniques + plaintes proches de celle de la patiente dans sa jeunesse Mère : 59 ans, diabète de type II Tante paternelle : plaintes proches Toxiques et médicaments Stop tabac lors de la grossesse, pas d’alcool Biofer®, Cerazette® (minipilule)
")
3
Clinique 65 Kg, 10/7,5 cm Hg, 60/minute REBV
Auscultation cardiaque et des vaisseaux du cou : N Marche et ballant des M.S. : N Station debout correctement tenue sans Romberg Serment, Mingazzini : pas de parésie 5 génuflexions sans difficulté, absence de Gowers Trophisme musculaire : N R.O.T. : N aux 4 membres Réflexes cutanés plantaires : en flexion X 2 Sensibilités, nerfs crâniens et épreuves cérébelleuses cinétiques : N

4
Clinique Réactions myotoniques à la percussion des muscles thénariens bilatéralement

5
E.N.M.G. EMG des muscles Ext dig com, Biceps, Tibialis ant dr
Repos : silence électrique Percussion musculaire et changements de position de l’aiguille : rares myotonies Act. Volontaire : discrets signes myogènes Neurographie motrice et sensitive : N

7
Exploration Bilan biologique
Bilan hémo-leucocytaire : éosinophilie augmentée Électrophorèse de l’Hb (ß-thalassémie) : N Bilan inflammatoire : N Ac anti-muscles striés et anti-canaux salivaires : + CPK et myoglobine : N Bilan lipidique et biochimie génétique : N K+ (paralysie périodique) : N Bilan hépatique : gamma GT augm. (58 UI/l) TSH : N
 : N. Bilan inflammatoire : N. Ac anti-muscles striés et anti-canaux salivaires : + CPK et myoglobine : N. Bilan lipidique et biochimie génétique : N. K+ (paralysie périodique) : N. Bilan hépatique : gamma GT augm. (58 UI/l) TSH : N.")
8
Absence de dystrophie myotonique de Steinert
Exploration Biologie moléculaire 5 à 30 répétitions du triplet CTG dans le gène codant pour la myotonine protéine-kinase, cAMP dépendante, situé sur le chromosome 19 Absence de dystrophie myotonique de Steinert

9
Myotonies Cliniquement : retard de relaxation musculaire après contraction volontaire ou percussion Sur le plan électrique : contraction musculaire prolongée, décharge répétitive Symptômes Physiopathologie ENMG Blocage Raideur Hyperexcitabilité membranaire Myotonie électrique Faiblesse Inexcitabilité membranaire et/ou composante dystrophique Réduction de la taille de la réponse M au repos ou après tests de provocation (froid, effort) Tracés myogènes Douleur
 Tracés myogènes. Douleur.")
10
Classification : Myotonies
Pathologies dystrophiques - Dystrophie myotonique (Steinert) - Myopathie myotonique proximale (PROMM) Pathologies non-dystrophiques - Canalopathies chlore Myotonie congénitale (AD Thomsen, AR Becker) - Canalopathies sodium Paramyotonie congénitale (Eulenburg)- Adynamie épisodique (Gamstorp) Myotonie fluctuante Myotonie permanente Myotonie sensible à l’acétazolamide Paramyotonie et adynamie épisodique ou paralysie hyperkaliémique ont longtemps été considérées comme des maladies distinctes. Pourtant, il s’agit probablement d’une même affection dont l’expression phénotypique est variable. Deux indices allant dans ce sens: dans les 2 cas il s’agit d’une mutation du gène codant pour la sous-unité alpha du canal Na dans certaines familles, certains membres souffrent d’une paramyotonie, tandis que d’autre présentent des crises d’adynamie. Les femmes souffriraient plus souvent de paramyotonie; l’excitabilité des f.m. pourrait dépendre des H sexuelles ou il pourrait exister un canal ionique stabilisateur de membrane dépendant du Chromosome X Déficience en maltase acide 17q23 Paralysie périodique hypokalièmique : pas de myotonie (rare au niveau des paupières) - Schwartz-Jampel (éléments malformatifs multiples, myotonies et pseudomyotonies)
 - Myopathie myotonique proximale (PROMM) Pathologies non-dystrophiques - Canalopathies chlore Myotonie congénitale (AD Thomsen, AR Becker) - Canalopathies sodium Paramyotonie congénitale (Eulenburg)- Adynamie épisodique (Gamstorp) Myotonie fluctuante Myotonie permanente Myotonie sensible à l’acétazolamide. Paramyotonie et adynamie épisodique ou paralysie hyperkaliémique ont longtemps été considérées comme des maladies distinctes. Pourtant, il s’agit probablement d’une même affection dont l’expression phénotypique est variable. Deux indices allant dans ce sens: dans les 2 cas il s’agit d’une mutation du gène codant pour la sous-unité alpha du canal Na. dans certaines familles, certains membres souffrent d’une paramyotonie, tandis que d’autre présentent des crises d’adynamie. Les femmes souffriraient plus souvent de paramyotonie; l’excitabilité des f.m. pourrait dépendre des H sexuelles ou il pourrait exister un canal ionique stabilisateur de membrane dépendant du Chromosome X. Déficience en maltase acide 17q23. Paralysie périodique hypokalièmique : pas de myotonie (rare au niveau des paupières) - Schwartz-Jampel (éléments malformatifs multiples, myotonies et pseudomyotonies)")
11
Myotonies acquises Myotonies induites Myotonies révélées
Clofibrate (Atromidin®) Inhibiteurs de l’hydroxymethylglutaryl-CoA-reductase (Lipitor ®, Lescol®, Pravasine®, Zocor®) Chloroquine (Nivaquine®, Plaquenil®) Colchicine 2,4-Dichlorophenoxyacetate (herbicide bloquant la gCl-) Myotonies révélées Suxamethonium (Myoplégine®) ß2 mimétiques: Fenoterol (Berotec®, Duovent®), Ritodrine (Pre-par®) Myotonies des neuropathies : syn. d’Hausmanowa-Petrusewicz Myotonies vraies, sporadiques, aux M. Sup. et à la langue Neuropathie périphérique 20-25 diazacholestérol : desmostérol qui s’accumule dans la membrane créant une anomalie de la gNa et de la gK
 Inhibiteurs de l’hydroxymethylglutaryl-CoA-reductase (Lipitor ®, Lescol®, Pravasine®, Zocor®) Chloroquine (Nivaquine®, Plaquenil®) Colchicine. 2,4-Dichlorophenoxyacetate (herbicide bloquant la gCl-) Myotonies révélées. Suxamethonium (Myoplégine®) ß2 mimétiques: Fenoterol (Berotec®, Duovent®), Ritodrine (Pre-par®) Myotonies des neuropathies : syn. d’Hausmanowa-Petrusewicz. Myotonies vraies, sporadiques, aux M. Sup. et à la langue. Neuropathie périphérique diazacholestérol : desmostérol qui s’accumule dans la membrane créant une anomalie de la gNa et de la gK.")
12
Dystrophie myotonique (Steinert, 1909)
Début : de 0 (forme congénitale) à l’âge adulte Myotonie : souvent méconnue, améliorée par la répétition du mouvement (warm-up), prédomine au niveau des mains Atrophie musculaire et faiblesse : face, cou (s.c.m.), segments distaux des membres Douleur : 0 Cataracte (lampe à fente), calvitie fronto-pariétale précoce cœur : BB, bloc AV, flutter et fibrillation auriculaire hypogonadisme, atrophie testiculaire, intolérance au glucose, diabète II, hypersomnie, apnées centrales, troubles de la personnalité, neuropathie périphérique… Diminution du potentiel de repos membranaire Diminution de la concentration en NaK ATPase Persistance anormale d’un canal K+ Ca++ dépendant entraînant un post potentiel hyperpolarisant qui favorise le réactivation rapide du canal sodium Le produit anormal du gène est la myotonine protéine kinase qui a une action membranaire par phosphorylation des canaux ioniques L’amplification est plus important en cas de transmission féminine. < 100 répétitions CTG l’amplification est sous influence paternelle 500 répétitions CTG l’amplification est sous influence maternelle Forme congénitale : NN de mère atteinte Symtômes cardiaques, risque de mort subite, stimulation cardiaque préventive Poumons : surv par EFR et gazométrie artérielle Effort court (10’’) : réduction de la M à T0 Génétique : AD, pénétrance complète, expressivité variable (anticipation), Ch 19q13.3 (1992), amplification d’une répétition du trinucléotide CTG (> 50) au niveau du gène codant pour la myotonine protéine kinase (DMPK) ENMG : myotonies électriques, tracés myogènes
 à l’âge adulte. Myotonie : souvent méconnue, améliorée par la répétition du mouvement (warm-up), prédomine au niveau des mains. Atrophie musculaire et faiblesse : face, cou (s.c.m.), segments distaux des membres. Douleur : 0. Cataracte (lampe à fente), calvitie fronto-pariétale précoce cœur : BB, bloc AV, flutter et fibrillation auriculaire hypogonadisme, atrophie testiculaire, intolérance au glucose, diabète II, hypersomnie, apnées centrales, troubles de la personnalité, neuropathie périphérique… Diminution du potentiel de repos membranaire. Diminution de la concentration en NaK ATPase. Persistance anormale d’un canal K+ Ca++ dépendant entraînant un post potentiel hyperpolarisant qui favorise le réactivation rapide du canal sodium. Le produit anormal du gène est la myotonine protéine kinase qui a une action membranaire par phosphorylation des canaux ioniques. L’amplification est plus important en cas de transmission féminine. < 100 répétitions CTG l’amplification est sous influence paternelle. 500 répétitions CTG l’amplification est sous influence maternelle. Forme congénitale : NN de mère atteinte. Symtômes cardiaques, risque de mort subite, stimulation cardiaque préventive. Poumons : surv par EFR et gazométrie artérielle. Effort court (10’’) : réduction de la M à T0. Génétique : AD, pénétrance complète, expressivité variable (anticipation), Ch 19q13.3 (1992), amplification d’une répétition du trinucléotide CTG (> 50) au niveau du gène codant pour la myotonine protéine kinase (DMPK) ENMG : myotonies électriques, tracés myogènes.")
13
Myopathie myotonique proximale (PROMM, 1994)
Début : de 8 à 50 ans, pas de forme congénitale Myotonie : souvent méconnue, aggravée par la chaleur (améliorée par le froid) Atrophie musculaire et faiblesse : segments proximaux des membres (hypertrophie des mollets) Douleur : douleur ou inconfort musculaire Cataracte, arythmie cardiaque, hypogonadisme, intolérance au glucose, hypersomnie, hyperintensité T2 de la substance blanche à l’IRM… Génétique : AD, Ch 3q21 (2001), amplification d’une répétition du quadruplet CCTG ( ) au niveau du gène codant pour la protéine ZNF9 (amplification faible) ENMG : myotonies électriques (parfois peu abondantes), tracés myogènes
 Atrophie musculaire et faiblesse : segments proximaux des membres (hypertrophie des mollets) Douleur : douleur ou inconfort musculaire. Cataracte, arythmie cardiaque, hypogonadisme, intolérance au glucose, hypersomnie, hyperintensité T2 de la substance blanche à l’IRM… Génétique : AD, Ch 3q21 (2001), amplification d’une répétition du quadruplet CCTG ( ) au niveau du gène codant pour la protéine ZNF9 (amplification faible) ENMG : myotonies électriques (parfois peu abondantes), tracés myogènes.")
14
Myotonie Congénitale:Thomsen AD (1876), Becker AR (1971)
Canalopathies Cl Myotonie Congénitale:Thomsen AD (1876), Becker AR (1971) Début : naissance-puberté (AD), 5-12 ans (AR) Myotonie d’intention diffuse, plus sévère distalement, aux M. Inf., chez l’homme et dans la forme récessive, warm-up Faiblesse musculaire (AR) : proximale, corrigée par la répétition de la contraction (warm-up) Hypertrophie musculaire : diffuse prédominant aux M.Inf. et dans la forme récessive (coexistant parfois avec une atrophie distale aux M.Sup.) Douleur : des myalgies sont parfois décrites Génétique : Ch 7q35, mutation du gène codant pour le canal chlore musculaire (CLCN1) responsable d’une réduction de la gCL induisant une instabilité avec hyperexcitabilité membranaire (myotonie) et/ou une inexcitabilité membranaire (faiblesse) Certaines mutations sont communes aux formes récessive et dominante ???? La diminution de la gCl fait que l’accumulation de K+ extracellulaire n’est plus compensée par l’entrée de Cl Le warm-up correspondrait à la mise en jeu de la NaK ATPase La myotonie peut être responsable d’un trouble de l’élocution, de la déglutition, de la mastication ou même d’une diplopie ENMG : myotonies électriques; dans la forme récessive : décrément de la réponse M lors de la SNR à 10 Hz (4’’) ou après un effort bref de contraction (10’’), tracés myogènes
, Becker AR (1971) Début : naissance-puberté (AD), 5-12 ans (AR) Myotonie d’intention diffuse, plus sévère distalement, aux M. Inf., chez l’homme et dans la forme récessive, warm-up. Faiblesse musculaire (AR) : proximale, corrigée par la répétition de la contraction (warm-up) Hypertrophie musculaire : diffuse prédominant aux M.Inf. et dans la forme récessive (coexistant parfois avec une atrophie distale aux M.Sup.) Douleur : des myalgies sont parfois décrites. Génétique : Ch 7q35, mutation du gène codant pour le canal chlore musculaire (CLCN1) responsable d’une réduction de la gCL induisant une instabilité avec hyperexcitabilité membranaire (myotonie) et/ou une inexcitabilité membranaire (faiblesse) Certaines mutations sont communes aux formes récessive et dominante La diminution de la gCl fait que l’accumulation de K+ extracellulaire n’est plus compensée par l’entrée de Cl. Le warm-up correspondrait à la mise en jeu de la NaK ATPase. La myotonie peut être responsable d’un trouble de l’élocution, de la déglutition, de la mastication ou même d’une diplopie. ENMG : myotonies électriques; dans la forme récessive : décrément de la réponse M lors de la SNR à 10 Hz (4’’) ou après un effort bref de contraction (10’’), tracés myogènes.")
15
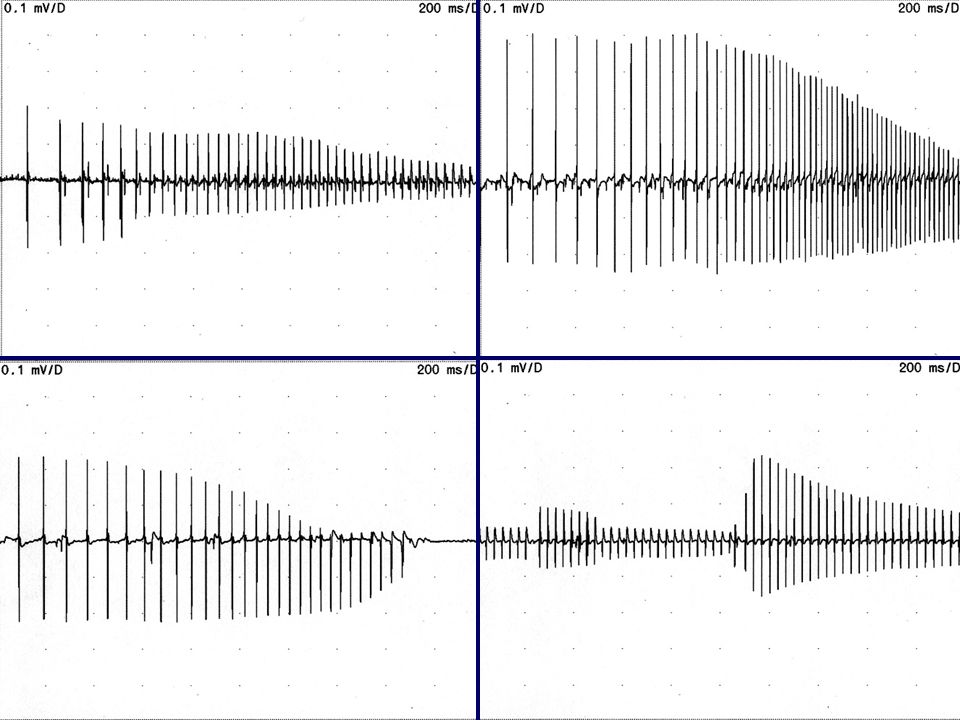
SNR 10 Hz pendant environ 4’’
En haut myotonie congénitale de type Becker En bas paralysie périodique (NA ou Ca)
")
16
EMG de surface au niveau du grand palmaire : 3X 4’’ à 30’’ d’interv
A gauche myotonie congénitale de type Becker : décrément et warm-up A droite paralysie périodique (Na ou Ca)
")
17
Paramyotonie congénitale (Eulenburg)-Adynamie épisodique (Gamstorp)
Canalopathies Na Paramyotonie congénitale (Eulenburg)-Adynamie épisodique (Gamstorp) Début : enfance, adolescence Myotonie paradoxale aggravée par l’exercice, par le froid, de topographie cheiro-facio-linguale (fermeture répétée des yeux) Faiblesse musculaire : accès paralytiques (dizaine de minutes à une heure ou plus) déclenchés par un court repos après un exercice important, le froid, le jeune, un repas riche en K+ Trophicité musculaire habituellement normale Douleur : 0 Génétique : AD, Ch 17q23-25, mutation du gène codant pour la sous-unité alpha du canal Na musculaire votage-dépendant (SCN4A) responsable d’un retard d’inactivation du canal induisant un gain de fonction avec instabilité et hyperexcitabilité membranaire (myotonie) ou inexcitabilité membranaire (faiblesse) Adynamie : potentiel de repos membranaire est diminué (-46 mV) en raison de l’augmentation de potassium extracellulaire + augmentation de la gNa par retard d’inactivation des canaux Na Paramyotonie : potentiel de repos membranaire est normal (-80 mV) à 37°C, mais la gNa augmente anormalement trop lors du refroidissement Dans certaines fammiles : myotonies aggravées par la chaleur (été) et adynamie liée au froid (hiver) avec hypokaliémie Entre les crises, il peut persister un certain degré de faiblesse proximale L’épisode d’adynamie est souvent annoncé par des paresthésies et la réalisation d’un effort peut éviter la crise ENMG : myotonies électriques (parfois uniquement au froid : 29°C); incrément (> 300%) après contraction volontaire (5’) pendant les crises et test de McManis en dehors des crises
-Adynamie épisodique (Gamstorp) Début : enfance, adolescence. Myotonie paradoxale aggravée par l’exercice, par le froid, de topographie cheiro-facio-linguale (fermeture répétée des yeux) Faiblesse musculaire : accès paralytiques (dizaine de minutes à une heure ou plus) déclenchés par un court repos après un exercice important, le froid, le jeune, un repas riche en K+ Trophicité musculaire habituellement normale. Douleur : 0. Génétique : AD, Ch 17q23-25, mutation du gène codant pour la sous-unité alpha du canal Na musculaire votage-dépendant (SCN4A) responsable d’un retard d’inactivation du canal induisant un gain de fonction avec instabilité et hyperexcitabilité membranaire (myotonie) ou inexcitabilité membranaire (faiblesse) Adynamie : potentiel de repos membranaire est diminué (-46 mV) en raison de l’augmentation de potassium extracellulaire + augmentation de la gNa par retard d’inactivation des canaux Na. Paramyotonie : potentiel de repos membranaire est normal (-80 mV) à 37°C, mais la gNa augmente anormalement trop lors du refroidissement. Dans certaines fammiles : myotonies aggravées par la chaleur (été) et adynamie liée au froid (hiver) avec hypokaliémie. Entre les crises, il peut persister un certain degré de faiblesse proximale. L’épisode d’adynamie est souvent annoncé par des paresthésies et la réalisation d’un effort peut éviter la crise. ENMG : myotonies électriques (parfois uniquement au froid : 29°C); incrément (> 300%) après contraction volontaire (5’) pendant les crises et test de McManis en dehors des crises.")
18
Canalopathies Na Myotonie fluctuante : début dans l’enfance d’une myotonie retardée survenant après l’exercice (qq minutes à 30’) parfois accompagnée de douleur, agravée par le K+, non aggravée par le froid, sans faiblesse musculaire, avec une trophicité musculaire normale Myotonie permanente : myotonie sévère permanente pouvant entraver la respiration, accompagnée d’une hypertrophie musculaire importante Myotonie sensible à l’acétazolamide : myotonie paradoxale non aggravée par le K+, hypertrophie musculaire La myotonie retardée dure environ 1 heure Génétique : Ch 17q23-25, mutation du gène codant pour la sous-unité alpha du canal Na musculaire votage-dépendant (SCN4A) ENMG : myotonies électriques
 parfois accompagnée de douleur, agravée par le K+, non aggravée par le froid, sans faiblesse musculaire, avec une trophicité musculaire normale. Myotonie permanente : myotonie sévère permanente pouvant entraver la respiration, accompagnée d’une hypertrophie musculaire importante. Myotonie sensible à l’acétazolamide : myotonie paradoxale non aggravée par le K+, hypertrophie musculaire. La myotonie retardée dure environ 1 heure. Génétique : Ch 17q23-25, mutation du gène codant pour la sous-unité alpha du canal Na musculaire votage-dépendant (SCN4A) ENMG : myotonies électriques.")
19
Exploration Biologie moléculaire (Paris, service du Dr B. HAINQUE) Absence de mutation du gène du canal sodium musculaire SCN4A. En particulier, absence des mutations décrites pour la myotonia fluctuans.

20
Contribution de l'ENMG au diagnostic des Myotonies
Myotonie : déclenchée ou aggravée par le froid (paramyotonie), parfois myotonies infracliniques chez un hétérozygote pour la forme récessive de la myotonie congénitale Tracés myogènes : Steinert, PROMM, Becker, durant les crises d’adynamie épisodique Incrément (> 300%) après contraction volontaire (5’) ou SNR prolongée (Abd. V) : pendant les crises d’adynamie épisodique Décrément (> 25 %) après SNR à 10 Hz (4’’) ou exercice court (contraction volontaire de 10’’): Myotonie congénitale (AR) Exercice long (5’ : contraction volontaire 25’’-repos 5’’) (McManis) Pendant ou juste après l’effort : incrément > 30% adynamie épisodique ou pp secondaire à une thyrotoxicose Après effort : décrément > 40% adynamie épisodique ou pp secondaire à une thyrotoxicose et PROMM
, parfois myotonies infracliniques chez un hétérozygote pour la forme récessive de la myotonie congénitale. Tracés myogènes : Steinert, PROMM, Becker, durant les crises d’adynamie épisodique. Incrément (> 300%) après contraction volontaire (5’) ou SNR prolongée (Abd. V) : pendant les crises d’adynamie épisodique. Décrément (> 25 %) après SNR à 10 Hz (4’’) ou exercice court (contraction volontaire de 10’’): Myotonie congénitale (AR) Exercice long (5’ : contraction volontaire 25’’-repos 5’’) (McManis) Pendant ou juste après l’effort : incrément > 30% adynamie épisodique ou pp secondaire à une thyrotoxicose. Après effort : décrément > 40% adynamie épisodique ou pp secondaire à une thyrotoxicose et PROMM.")
21
Secondary causes of periodic paralysis
Hypokalemic Hyperkalemic Thyrotocic Primary hyeraldosteronism (Conn) Urinary potassium wastage Renal tubular acidosis Juxtaglomular apparatus hyperplasia (Barter) Fanconi’s syndrome Gastrointestinal potassium wastage Villous adenoma Laxative abuse Pancreatic noninsulin-secreting tumors with diarrhea Nontropical sprue Barium intoxication Drug-induced Potassium-depleting diuretics Addison’s disease Hypoaldosteronism Excessive potassium supplementation Potassium-sparing diuretics Chronic renal failure Rhabdomyoloysis
 Urinary potassium wastage. Renal tubular acidosis. Juxtaglomular apparatus hyperplasia (Barter) Fanconi’s syndrome. Gastrointestinal potassium wastage. Villous adenoma. Laxative abuse. Pancreatic noninsulin-secreting tumors with diarrhea. Nontropical sprue. Barium intoxication. Drug-induced. Potassium-depleting diuretics. Addison’s disease. Hypoaldosteronism. Excessive potassium supplementation. Potassium-sparing diuretics. Chronic renal failure. Rhabdomyoloysis.")
22
sans effet dans la paramyotonie
Traitements Quinine (200 to mg/j) Phénytoïne ( mg/j) Méxilétine (150 to mg/j : Mexitil (antiarythmique de classe I, stabilisateur de membrane) Tocaïnide (toxicité hématologique) Dilantine (300 to 400 mg/j, niveau thérapeutique : µl/ml) Diurétiques thiazidiques (diminution du K+ extracellulaire) Procaïnamide (125 to mg/d) (risque de LE disséminé) Carbamazépine : Tegretol (inhibiteur du canal Na) Acétazolamide (125 to 750 mg/j) : Diamox (inhibiteur de l’anhydrase carbonique) diminution de la kaliémie par acidose métabolique) sans effet dans la paramyotonie
 Phénytoïne ( mg/j) Méxilétine (150 to mg/j : Mexitil (antiarythmique de classe I, stabilisateur de membrane) Tocaïnide (toxicité hématologique) Dilantine (300 to 400 mg/j, niveau thérapeutique : µl/ml) Diurétiques thiazidiques (diminution du K+ extracellulaire) Procaïnamide (125 to mg/d) (risque de LE disséminé) Carbamazépine : Tegretol (inhibiteur du canal Na) Acétazolamide (125 to 750 mg/j) : Diamox (inhibiteur de l’anhydrase carbonique) diminution de la kaliémie par acidose métabolique) sans effet dans la paramyotonie.")
23
Génétique des canalopathies Cl
Tableau clinique de myotonie congénitale si > 50% des canaux Cl- sont non fonctionnels Canal Cl- a une structure tétramérique 1 allèle muté et 1 allèle non muté : 4 monomères normaux : 1/ monomères anormaux : 1/ monomères normaux et 1 anormal : 4/ monomères anormaux et 1 normal : 4/ monomères normaux et 2 anormaux : 6/16 - Mutation qui rend le canal Cl- non fonctionnel quand 3 monomères sont anormaux : 5/16 des canaux Cl- sont non-fonctionnels Hétérozygote de la forme AR (myo. infraclinique chez l’homme) - Mutation qui rend le canal Cl- non fonctionnel quand 1 ou 2 monomères sont anormaux : 15/16 ou 10/16 des canaux sont non-fonctionnels Forme AD 2 allèles mutés : 100% des canaux Cl- sont non fonctionnels Homozygote de la forme AR (forme plus sévère que l’AD)
 - Mutation qui rend le canal Cl- non fonctionnel quand 1 ou 2 monomères sont anormaux : 15/16 ou 10/16 des canaux sont non-fonctionnels Forme AD. 2 allèles mutés : 100% des canaux Cl- sont non fonctionnels Homozygote de la forme AR (forme plus sévère que l’AD)")
Présentations similaires






 Pr E. Tournier-Lasserve>")












