Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
UE Méthodes d'étude et analyse du génome
Pré-requis cours de Biochimie relatif aux acides nucléiques et aux protéines. Structure ADN , ARN

2
I-Extraction et Purification de l’ ADN
L’étude du génome commence par une extraction suivie d’une purification de l’ADN Sources cellulaires d’ADN : Noyau , Mitochondries Suppose fragmentation cellulaire Les Globules rouges n’ont pas d’ADN …

3
Méthode de référence : Phénol/chloroforme
Sélection de cellules : Leucocytes Lyse Membranaire : Lysozyme, SDS et/ou autre détergeant ionique en phase aqueuse à pH neutre Dans l’ordre Eliminer ARN : Rnase (s) Eliminer protéines : Protéase(s) -incubationplusieurs heures à 37° c
 Eliminer protéines : Protéase(s) -incubationplusieurs heures à 37° c.")
4
Action du phénol et du chloroforme pour éliminer les macromécules non ADN de l’extrait cellulaire et solubilisation de l’ADN en phase aqueuse Par centrifugation , les autres molécules sont éliminées de l’interface Eau/ Phénol-chloroforme (X 2, 3)
")
5
L’ addition d’ alcool isoamylique élimine le phénol en excès
On ajoute à la phase aqueuse , une solution saline concentrée

6
Précipitation sélective de l'ADN en présence de 0. 25-0
Précipitation sélective de l'ADN en présence de M sel ionisable ( Acétate d’ammonum, NaCl) Par exemple Verser doucement 2V acétate de sodium dans le tube contenant l'ADN Agiter, Puis ajouter 3 V d’éthanol froid . Agiter. L'ADN précipite alors sous forme de filaments blanchâtres formant la “ méduse ”. On peut recueillir ensuite cet ADN en l'enroulant autour de l'extrémité d'une pipette fine et le mettre en solution dans 0.5 à 1 ml de tampon contenant 1à 2mM d’EDTA. (TE)
 Par exemple Verser doucement 2V acétate de sodium dans le tube contenant l ADN Agiter, Puis ajouter 3 V d’éthanol froid . Agiter. L ADN précipite alors sous forme de filaments blanchâtres formant la méduse . On peut recueillir ensuite cet ADN en l enroulant autour de l extrémité d une pipette fine et le mettre en solution dans 0.5 à 1 ml de tampon contenant 1à 2mM d’EDTA. (TE)")
8
2- Hybridation Moléculaire d’acides Nucléiques
L'hybridation moléculaire des acides nucléiques est à la base de nombreuses techniques d’exploration du Génome impliquant la mise en présence d'au moins deux simples brins d'acides nucléiques dans des conditions physico chimiques précises. Domine les techniques de l’étude du génome

9
L’hybridation moléculaire sert à la détection spécifique de séquences d’acides nucléiques pour des études d’expression de gènes, de leur séquence pour la détection de mutations , de polymorphismes etc… Applications importantes en diagnostic moléculaire des maladies génétiques

10
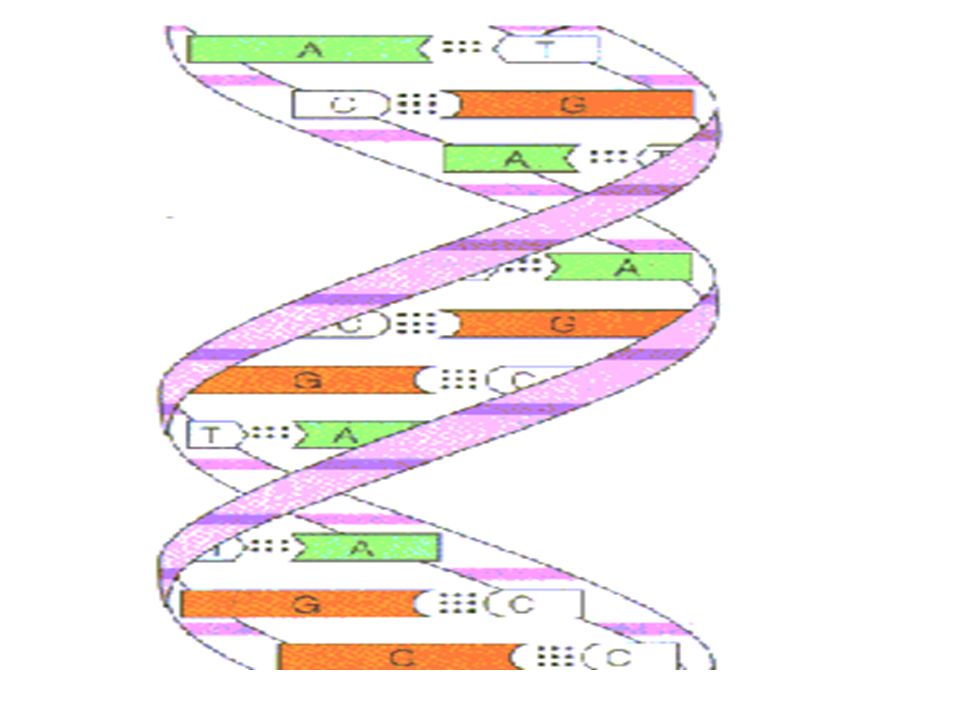
Définition L'hybridation moléculaire désigne l'association entre deux séquences d’ acides nucléiques simples brins complémentaires aboutissant alors à la formation d'un double brin ou duplex.

11
Cette association s'effectue par l’intermédiaire de liaisons hydrogènes spécifiques : deux liaisons entre l'adénine (A) et la thymine (T) (ou l'uracile U) et trois entre la cytosine (C) et la guanine (G). La formation et la stabilité des duplex dépendent de nombreux facteurs : Composition en bases Longueur du duplex formé La répétition des nucléotides Du milieu ou du support dans ou sur lequel l’hybridation aura lieu.

13
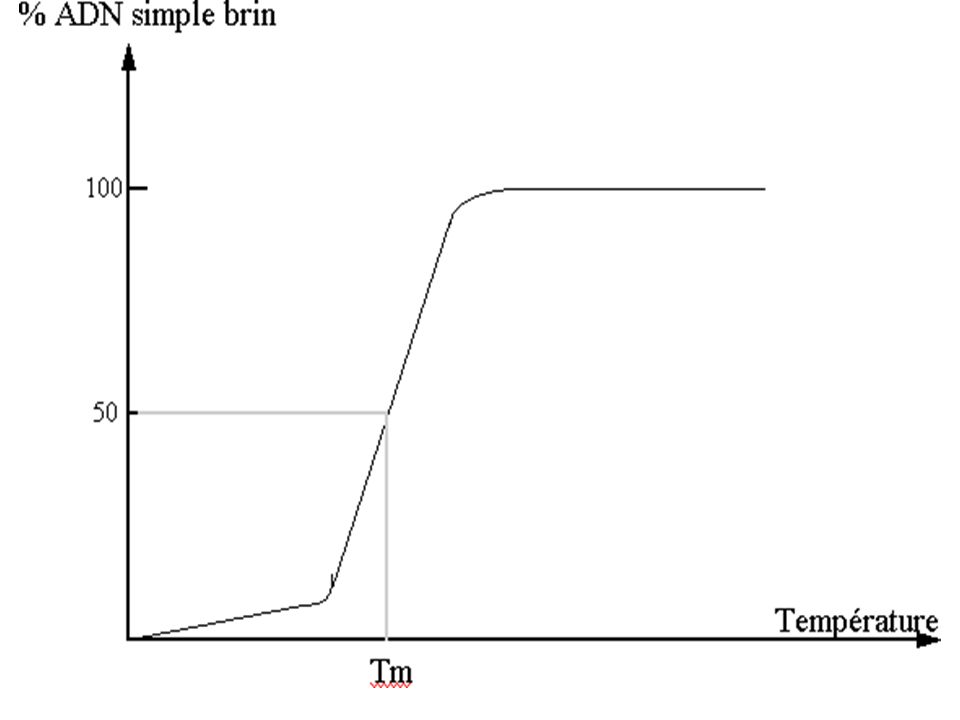
Bases de L’hybridation – TM
La température de fusion de l’ADN : est une température appelée également Température de demi dénaturation : Ou Tm qui correspond à l’ouverture de 50% de la double chaîne de l’ADN chauffé ( dénaturé)
")
14
C'est l'effet hyperchrome qui correspond à la rupture des liaisons hydrogènes (liaisons faibles)
La séparation des 2 chaînes entraîne une augmentation de l'absorption dans le spectre U.V à 260 nm. La valeur de Tm des ADN est une fonction linéaire du pourcentage de (G + C) de l'ADN :
 de l ADN :")
15
Le résultat du passage de la forme bicaténaire à la forme monocaténaire de l’ADN est une augmentation du coefficient d’extinction de l’ADN : c’est l’effet hyperchrome. Les bases azotées qui absorbent à cette longueur d’onde sont très ordonnées dans la forme bicaténaire .

16
Les bases azotées de l’ADN simple brin sont moins masquées et n’ont plus de structure ordonnée ( « quenching ») Le coefficient d’extinction moléculaire est plus élevé.

18
Calcul Du TM Pour moins de 14 nucléotides
Tm= (wA+xT) 2 + (yG+zC) 4 w,x,y,z nombre de A,T,G,C dans la séquence
 2 + (yG+zC) 4. w,x,y,z nombre de A,T,G,C dans la séquence.")
19
Séquences longues Tm= (yG+zC16.4)/(wA+xT+yG+zC)
/(wA+xT+yG+zC)")
20
Application à l’étude des Génomes

21
- Composition du génome humain en fonction du type de séquences
Coupure a a’ a Dénaturation a’

22
- Réassociation ou Renaturation
Conditions choc entre a et a’ (concentration) Taux de renaturation = f( ) du carré de la concentration en simple brin dc dt = - kC2 c = [S. Brin] moles de nucléotides par litre t = sec k = cste 2ème : litres /seconde /moles
 Taux de renaturation = f( ) du carré de la concentration en simple brin. dc. dt. = - kC2. c = [S. Brin] moles de nucléotides par litre. t = sec. k = cste 2ème : litres /seconde /moles.")
23
A t = o c = Co 1 C Co - = kt C 1 = « Cot » Co 1 + kCot fraction S. Brin dans une cinétique de renaturation au temps t est une fonction de la [C initiale en S.B.] Co par temps Cot

24
Le brin, dont on connaît au moins une partie de la séquence, est une sonde, l'autre brin, celui que l'on souhaite caractériser constitue la cible. L'un des deux brins est marqué par couplage chimique avec une molécule pouvant générer un signal.

25
S I M P L E B R N Cot (mole sec .l-1) Rapide Lent 100
ADN hautement répétitif S I M P L E B R N % 60 ADN moyennement répétitif ADN peu répétitif 20 10-2 Copies uniques 10-3 100 101 102 104 Cot (mole sec .l-1) Rapide Lent Taux de Renaturation
 Rapide. Lent. Taux de Renaturation.")
26
Plus le nombre de séquences complémentaires est grand plus le « Cot est petit » le nombre de séquences répétées va influencer la valeur du Cot [s. répétées] CoT

27
Plus le nombre de séquences complémentaires est grand plus le « Cot est petit » le nombre de séquences répétées va influencer la valeur du Cot [s. répétées] CoT

28
3- Caractéristiques du génome humain
Par cellule / génome haploïde 3,3 109 pb MM = 1,8 1012 daltons Taille d'un gène 100 pb 2,6 millions Quantité ADN haploide / cellule : 3 picogrammes

29
4- Organisation des séquences du Génome

30
ADN HAUTEMENT REPETITIF
L'ADN hautement répété n'est pas codant. Il représente 15 à 20% du génome. Il est constitué de séquences hétérochromatiques donc localisées principalement autour des centromères. Cet ADN hautement répété comprend : des répétitions de motifs courts (5 à 10 bp) disposés en tandem et répétés jusqu'à plusieurs centaines de milliers de fois;
 disposés en tandem et répétés jusqu à plusieurs centaines de milliers de fois;")
31
des séquences répétées de motifs toujours disposés en tandem mais plus longues (100 à 200 bp)
des séquences hautement répétées et dispersées dans tout le génome, les micro satellites ( en tandem également)
")
32
les centromères dans la région desquels on reconnaît des séquences CEN qui chez l'homme sont des motifs de 171 bp répétées en tandem dont la longueur varie de 300 kpb à kbp ; on les rencontre identiques sur tous les chromosomes

33
les telomères et séquences TEL situées à l'extrémité des chromosomes constituées de motifs répétés riches en C et A. Ces télomères auraient pour rôle de protéger l'extrémité des chromosomes de l'érosion au cours des réplications successives et de l'attaque des nucléases intracellulaires.

34
ADN hautement répété 105 copies
DNA satellite Simple a b (7 pb) n Télomères simple, a b Centromère (171) pb, n - Rôle architectural - C.O ADN non codant
 n Télomères simple, a. b Centromère (171) pb, n. - Rôle architectural. - C.O. ADN non codant.")
35
ADN hautement répétitif en tandem
ADN satellite Satellite Taille Nb copies Localisation Simple 5 - 25 ? Hétérochromatine : 1q, 9q, 16q, Yq a 171 8 x 105 Centromères b Sau3A family 68 5 x 104 9q, 13q, 14q, 15q, 21q, 22q Ces séquences ne sont pas transcrites

36
Satellite simple [ATTGCATTCCATATC] x n
![Satellite simple [ATTGCATTCCATATC] x n](http://slideplayer.fr/slide/9480482/29/images/36/Satellite+simple+%5BATTGCATTCCATATC%5D+x+n.jpg "Satellite simple [ATTGCATTCCATATC] x n")
37
Famille des minisatellites
taille Copies Localisation Famille télomérique 6 2-3 x 104 Télomères Famille hypervariable (pré-télomériques) 9 - 64 3 x 104 Tous les chromosomes + tous les télomères Famille HV [GGGCAGGA X G] 0,1 à 20 kb
 x 104. Tous les chromosomes + tous les télomères. Famille HV. [GGGCAGGA X G] 0,1 à 20 kb.")
38
ADN Moyennement Répété Séquences répétées dispersées

39
Eléments transposables
IS éléments 2600 – 700 pb 5' 3' CTGACTT 3' 5' TTCAGTC Répétition aux extrêmités (Maïs)
")
40
Séquences lines (Long interspersed repeat sequences)
pb Riches en AT 3' Peuvent contenir ORF Origine : transposition 20000 / copies L1 (KPn) 6,4 kb Repeat ORF1 ORF2 AAAA(A)n 6 kb Open reading frame
 6,4 kb. Repeat. ORF1. ORF2. AAAA(A)n. 6 kb. Open reading frame.")
41
Séquences SINEs 300 pb ALU REPEAT 500 000 copies
1 copie tous les 6 kb 3.6 % Génome 120 135 290 (AAA)n AT RICH GC RICH GC RICH ALU (AAA)n (AAA)n 130 31 290
n. AT RICH. GC RICH. GC RICH. ALU. (AAA)n. (AAA)n")
42
Sines = 5 % génome 1 séquence sine tous les 5000 pb (6000 pb pour ALU) ARNs transcrits à partir des séquences ALU mais pas de protéine traduite de ces ARNs

43
Les Microsatellites ( CA) n (CG) n Leur analyse détermine les empreintes génétiques
 n (CG) n Leur analyse détermine les empreintes génétiques")
44
Gène Séquence d'acides nucléiques contenant une information codée pour la production régulée d'un ARN (transcription), ce dernier pouvant être traduit en une chaîne polypeptique
, ce dernier pouvant être traduit en une chaîne polypeptique.")
45
Les gènes des rétrovirus sont constitués d'ARN

46
Pseudogènes Séquences partiellement homologues aux gènes qui ne donnent jamais de protéines correspondantes Soit anciens gènes fonctionnels qui ont muté Soit des ARN retro transcrit ADN Iintégration ADN génomique

47
1 30 31 104 105 146 IVS1 IVS2 Gène b globine 1600 pb Exon I Exon II
Zone des promoteurs 5' UTR 3' UTR 1 30 31 104 105 146 Exon I IVS1 Exon II IVS2 Exon III Gène b globine 1600 pb

48
Région régulatrice en 5' Zone des promoteurs
Facteurs de transcription -105 -100 -80 -70 CAP CACCC CACCC CCAAT TATA A/T A A/T A G G CCCCC Site d'initiation de la transcription

49
Région 5' non Traduite 5' UTR
CAP Codon initiateur CACCATG +50 CTTCTG Région 5' UTR - Attachement aux ribosomes - Régulation transcription +/-

50
Le CAPPING Les ARN messagers (ARNm) des cellules eucaryotes subissent d’importantes modifications avant d’être ultérieurement traduits en protéines. Dont l’ajout d’une structure-coiffe à l’extrémité 5’ des ARNm qui augmente considérablement la stabilité de ces molécules et promeut également une initiation efficace de la synthèse protéique.

52
Exon Partie codante d'un gène protéine Exon 1 ATG IVS1 30
Exons Traduction protéine

53
Zone non traduite IVS1 130 pb Exon 2 GT Donneur de l'épissage AG
Accepteur de l'épissage

54
Région 3' UTR Codon 147 146 AATAAA STOP 20 AAAAAA 132 Nucléotides
AATAAA : Signal de polyadénylation : Clivage de l'ARN après transcription (AAAAA)n : Stabilise l'ARN
n : Stabilise l ARN.")
55
Les sondes nucléotidiques
Définition Une sonde nucléotidique est un segment de nucléotides qui permet de rechercher de manière spécifique un fragment d'acide nucléique que l'on désire étudier et qui est complémentaire a la séquence de la sonde Cette réaction sonde-fragment correspond à une réaction d'hybridation moléculaire.

56
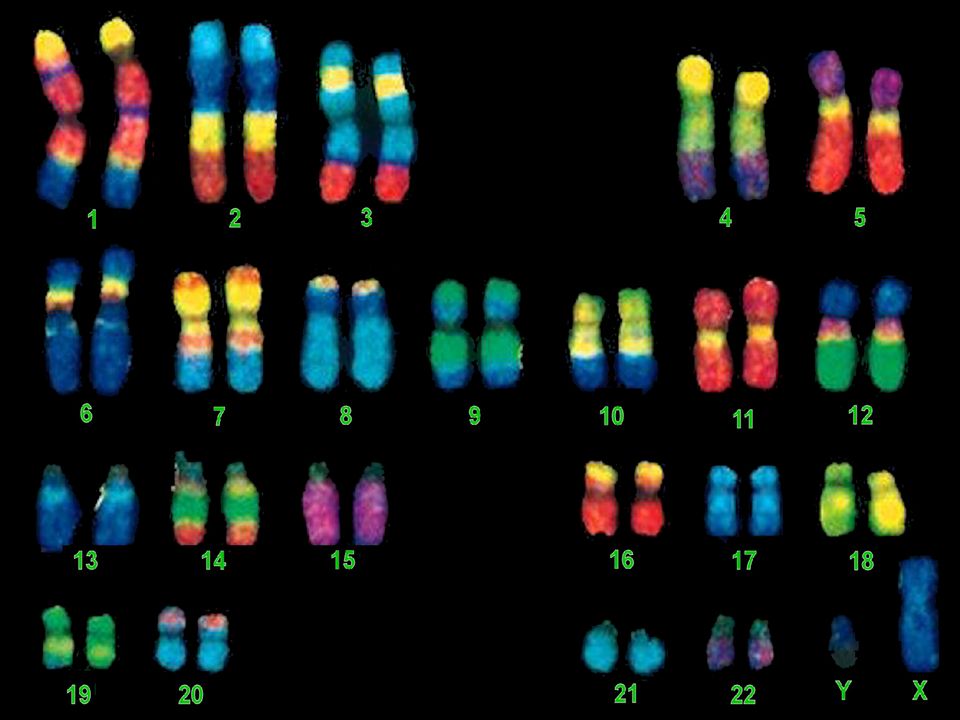
Analyse des Chromosomes
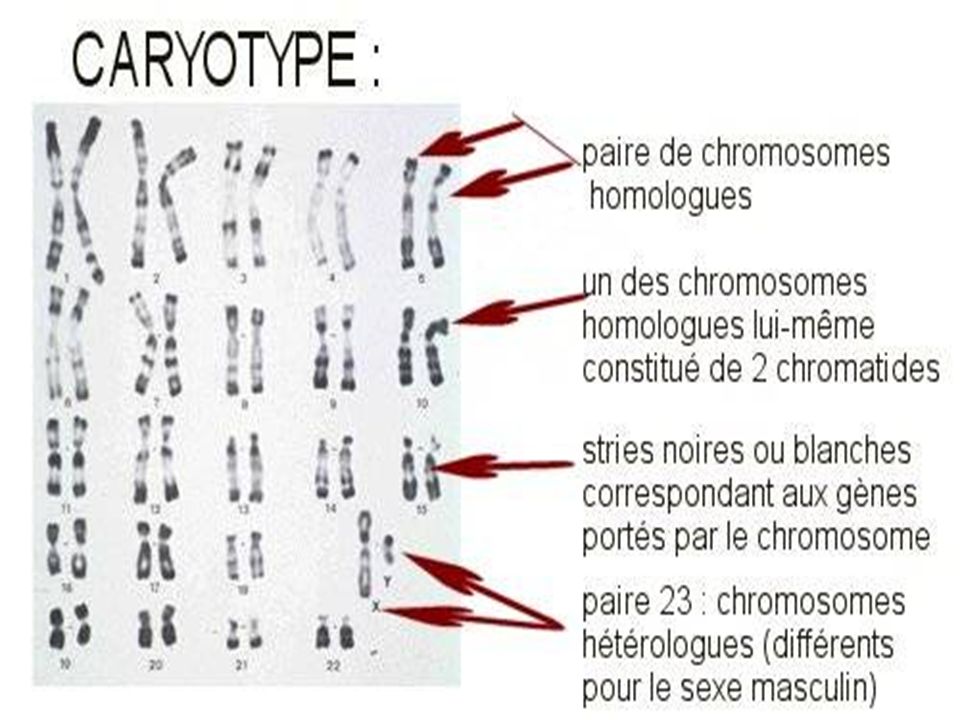
I- Le CARYOTYPE Classique

57
Le caryotype regroupe l'ensemble des chromosomes d'une cellule réunis par paires de chromosomes identiques et classés selon certains critères après une analyse dite de cytogénétique. En génétique médicale, le caryotype contribue à la mise en évidence de remaniements chromosomiques équilibrés ou déséquilibrés. La résolution d’un caryotype standard est celle d’une bande chromosomique, soit environ 5 à 10 millions de paires de bases Il sert à vérifier le nombre des chromosomes , leur morphologie , leur intégrité . Sa limite d’interprétation est liée à la taille des remaniements observables en microscopie . .

58
Tout prélèvement dont les cellules sont en division in vitro permet d’établir un caryotype
Il est nécessaire obtenir des métaphases nombreuses et de bonne qualité

59
Technique Stade de la culture cellulaire
Par ex: Le sang total est incubé 48 à 72 heures dans un milieu de culture: contenant une lectine à fort pouvoir mitogène (phytohémagglutinine ou PHA) permettant : une stimulation de la croissance des lymphocytes T.
 permettant : une stimulation de la croissance des lymphocytes T.")
60
* Blocage des cellules en métaphase: Colchicine
(empêche la polymérisation des tubulines donc la formation du fuseau mitotique) Lyse cellulaire contrôlée Libération des constituants cellulaires Fixation et étalement sur lame Les chromosomes libérés restent groupés Les lames sont ensuite observées par plusieurs dispositifs
 Lyse cellulaire contrôlée. Libération des constituants cellulaires. Fixation et étalement sur lame. Les chromosomes libérés restent groupés. Les lames sont ensuite observées par plusieurs dispositifs.")
61
Identification des chromosomes
Exemple / Par coloration au Giemsa Le marquage ou banding révèlent le long des chromosomes une alternance de bandes transversales, faiblement ou fortement colorées, dont la disposition topographique est spécifique de chaque paire chromosomique. Les bandes G (GTG) obtenues par dénaturation enzymatique et les bandes R (RHG) par dénaturation thermique Elles ont chacune un contenu spécifique en ADN. On obtient ainsi un repérage complémentaire
 obtenues par dénaturation enzymatique et les bandes R (RHG) par dénaturation thermique. Elles ont chacune un contenu spécifique en ADN. On obtient ainsi un repérage complémentaire.")
62
Un marquage en bandes G ou R permet de visualiser 300 à 600 bandes par lot haploïde de chromosomes.
Il existe des colorations spécifiques pour des régions précises. (ex : régions contenant les gènes codant les ARN des ribosomes) L’étude des cellules en prométaphase permet d’observer jusqu’à 800 bandes
 L’étude des cellules en prométaphase permet d’observer jusqu’à 800 bandes.")
63
Classification des chromosomes
Les chromosomes sont classés en fonction de leur taille décroissante et leur indice centromérique. Les techniques de marquage en bandes aident à identifier chaque paire chromosomique par le motif des bandes claires et sombres. Les bandes sont répertoriées dans une nomenclature internationale. Chaque bras chromosomique est divisé, selon sa taille, en une à quatre régions; chaque région en bandes numérotées du centromère vers le télomère

64
Par exemple la dénomination 6p12 signifie que la région est située sur le bras court du chromosome 6 et qu’il s’agit de la deuxième bande de la région 1

66
LIMITES La résolution d’un caryotype standard est celle d’une bande chromosomique, soit environ 5 à 10 millions de pb

67
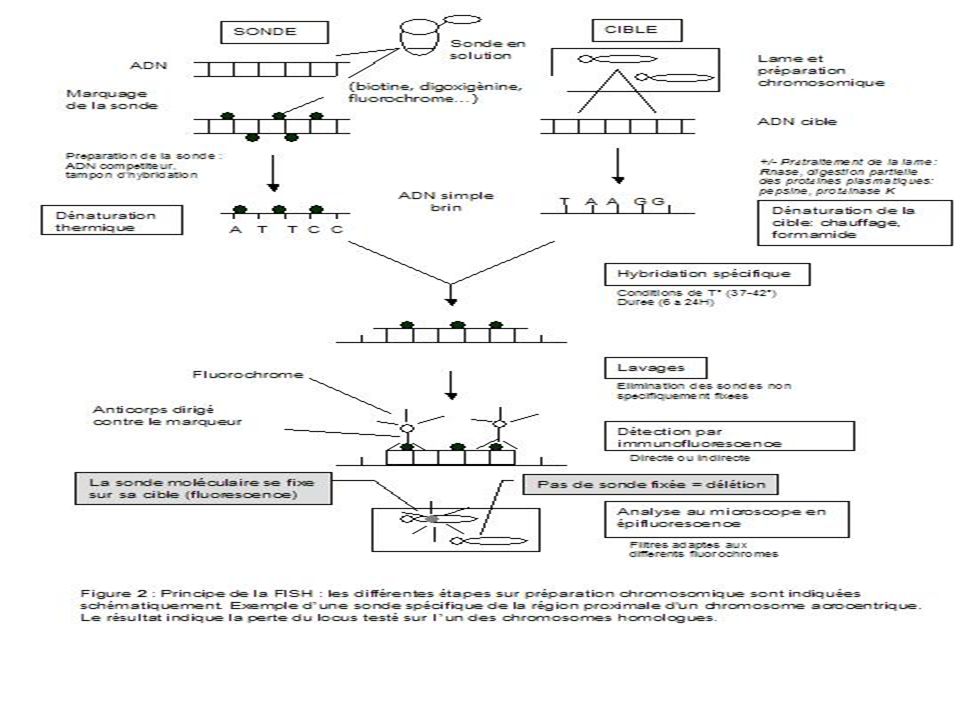
II- FISH ( Fluorescence in situ hybridization )
On utilise des sondes nucléiques dont la spécificité est variable . Elles peuvent être utilisées conjointement en grand nombre Elles sont fluorescentes en présence d’un dispositif d’observation spécifique

68
La FISH interphasique permet de s’affranchir de la culture cellulaire. Les signaux d’hybridation générés par les sondes d’une taille de plus de 150 kb sont visibles sur les noyaux. Un plus grand nombre de cellules (les noyaux sont 10 à 50 fois plus nombreux que les métaphases sur une préparation de cellules cultivées) peuvent être ainsi étudiées.
 peuvent être ainsi étudiées.")
70
Exemple Le chromosome Philadelphie est un chromosome chimérique : un chromosome créé par le pontage entre deux fragments d'ADN non contigus, l'un provenant d'une partie du chromosome 9 et l'autre d'une partie du chromosome 22. Translocation

71
La sonde verte indique la présence du gène BCR la sonde rouge indicate la présence du gène ABL . La fusion vert-rouge montre une fusion des 2 gènes .Ceci représente une translocation

73
Applications Parmi les principales applications, on peut citer le diagnostic prénatal des principales aneuploïdies, le diagnostic pré - implantatoire et celui des anomalies chromosomiques associées aux cancers.

74
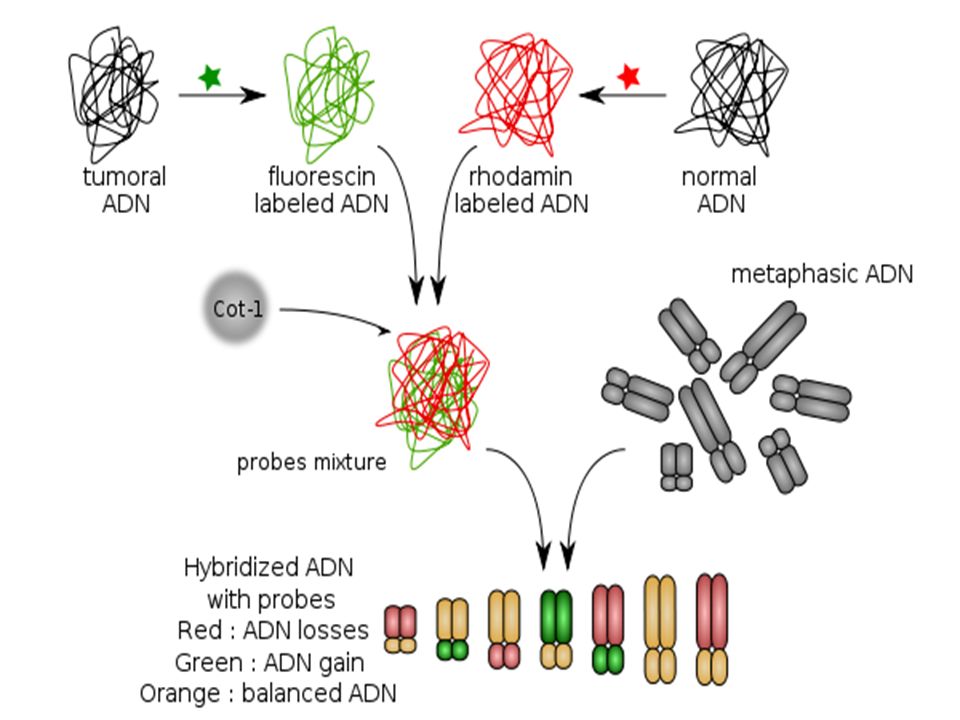
III- La CGH ( ( Hybridation génomique comparative)
C’est une technique basée sur l’ hybridation du génome entier d’un individu par rapport à un génome « normal » de référence Ces méthodes sont regroupées sous le vocable « caryotype moléculaire » et comprennent la technique des puces à ADN ou hybridation génomique comparative sur microréseau d'ADN.

75
Avec des chromosomes témoins
Avec des oligonucléotides

76
Chez l’homme Les Chromosomes sont présents en 2 exemplaires de chromosomes homologues
Dans certaines pathologies, le nombre de copies peut varier : il peut augmenter par exemple en cas de duplication, triplications … Et diminuer dans le cas de délétion. L'hybridation génomique comparative permet de diagnostiquer ces pathologies et d'identifier les régions chromosomiques impliquées.

77
III-a Avec chromosomes témoins
Principe L’ ADN de l’individu est marqué en vert L’ ADN témoin en rouge Des chromosomes « témoins » en métaphase sont fixés sur un support

79
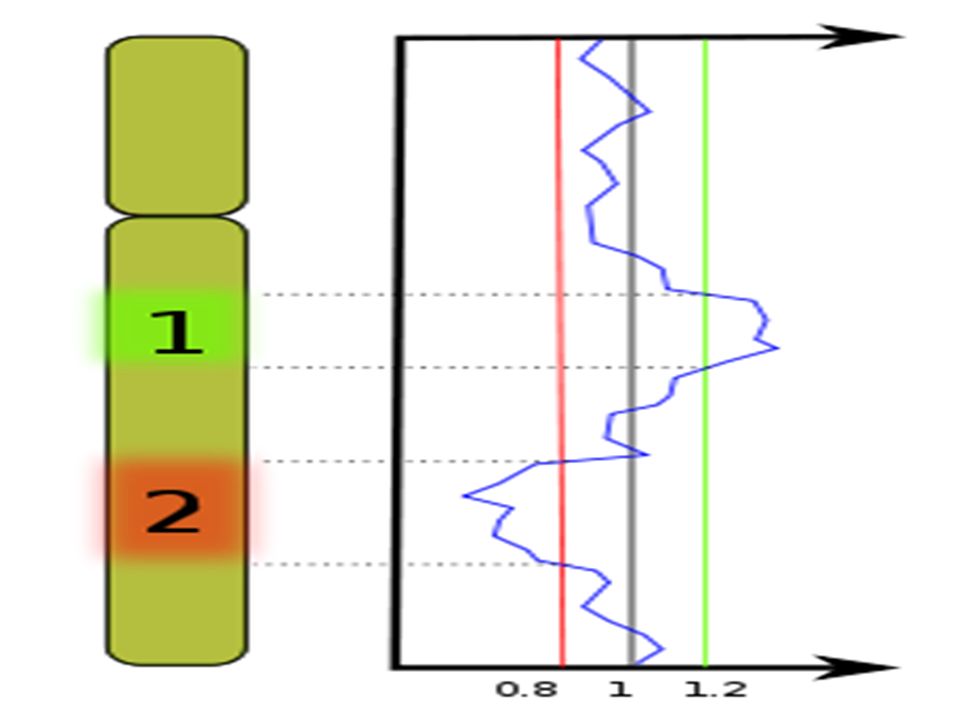
Exemple d’utilisation dans les cancers
si le nombre de copies est augmenté, la sonde tumorale s'hybridera préférentiellement et le segment de chromosome apparaitra en vert. si le nombre de copies est diminué, la sonde de tissu sain s'hybridera préférentiellement et le segment de chromosome apparaitra en rouge. si le nombre de copies n'est pas modifié, les deux sondes s'hybrideront en quantité équivalente et le segment de chromosome apparaitra en orangé.

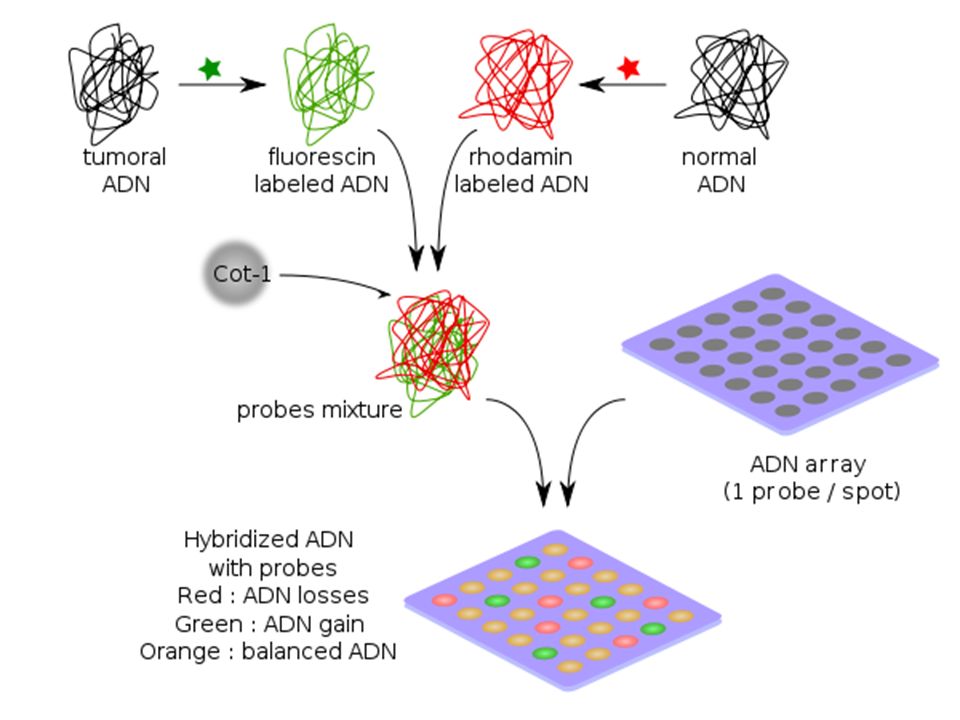
80
III-b Extension résolutive: Puces à ADN
Si l’on remplace les chromosomes en métaphase par des séquences d’ADN fixées sur le support et plus ou moins longues …ceci permettra d'augmenter la sensibilité, la gamme dynamique (la longueur des segments analysables) et le débit. La résolution augmente
 et le débit. La résolution augmente.")
83
Limites d’Utilisation
Cette technique détecte seulement les modifications chromosomiques non équilibrées. Des aberrations chromosomiques structurales telle que les translocations réciproques ou les inversions ne seront pas détectées.( contrairement au caryotype) Des délétions ou des duplications, des triplications de gènes seront détectées facilement
 Des délétions ou des duplications, des triplications de gènes seront détectées facilement.")
84
Séparation des Acides nucléiques
Par Electrophorèse Par Chromatographie

85
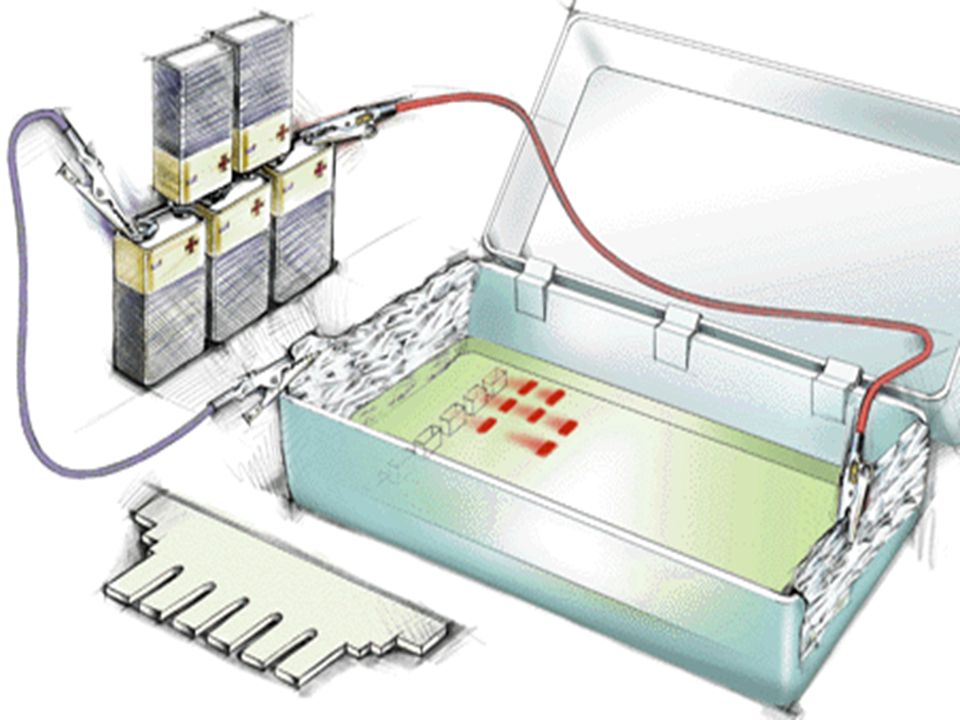
Electrophorèse des AN 2 supports ; Agarose; Acrylamide
A pH Neutre les AN sont chargés négativement, dans un champ électrique la migration s’effectue du pôle négatif vers le pôle positif Le critère de migration est la taille des fragments car la densité de charge /unité de longueur est constante. ( différence avec les protéines)
")
86
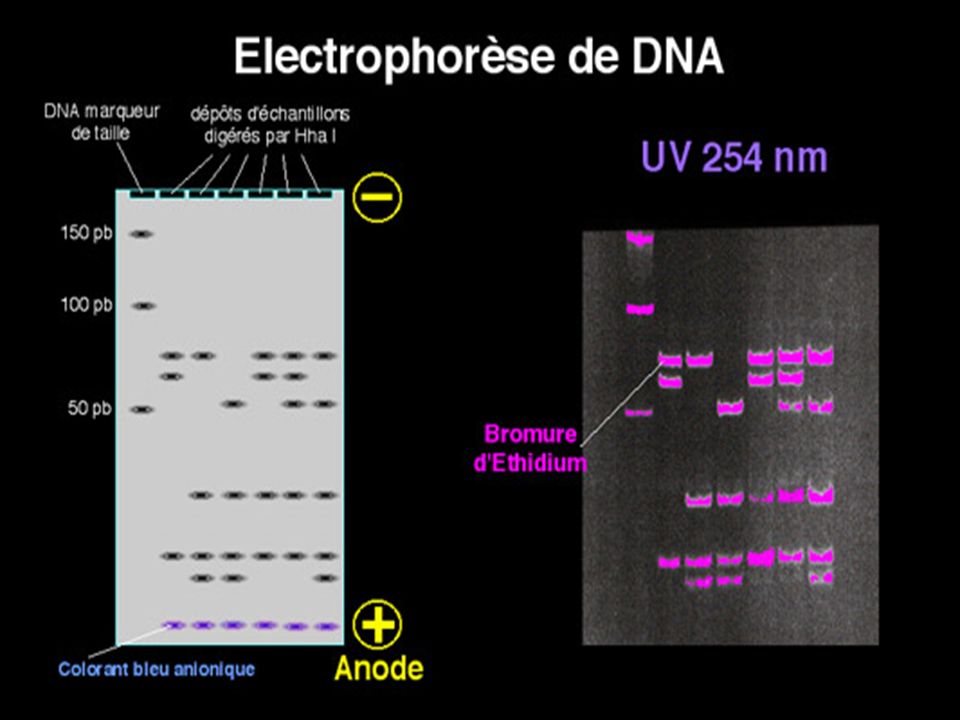
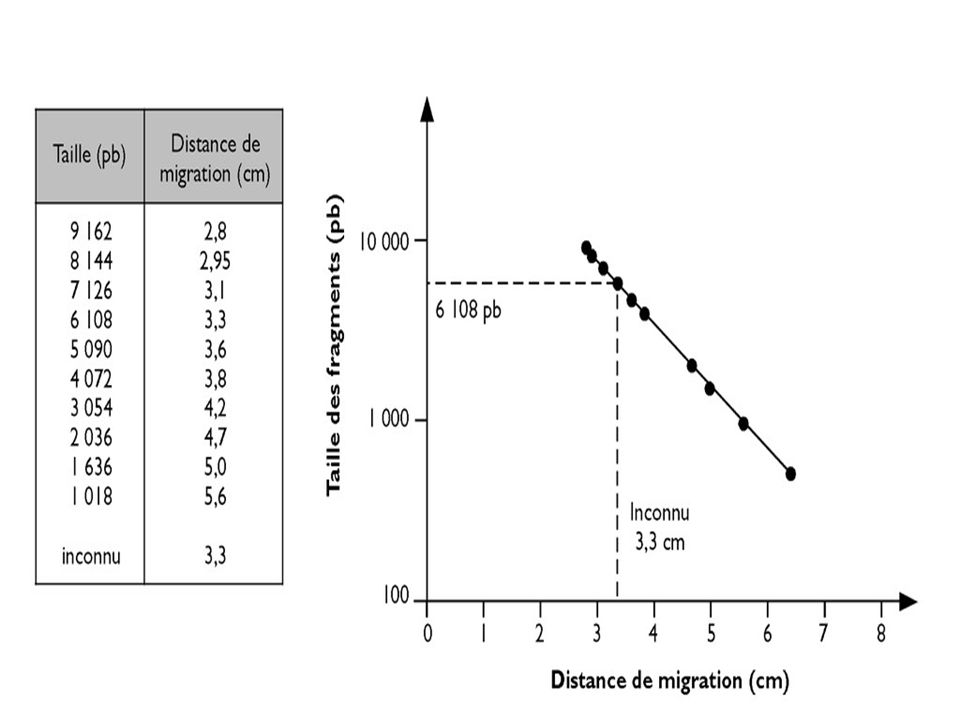
L’ADN doit être préalablement hydrolysé par un enzyme de restriction permettant d’obtenir des bandes distinctes si la masse moléculaire de l’ADN est petite ( < ou ~ pb) UN ADN génomique humain hydrolysé par un ER donnera un « Smear » ( très nombreux fragments donnant un aspect continu) On place dans des puits de dépôt des fragments de DNA de tailles connues pour servir de marqueurs de taille .
 On place dans des puits de dépôt des fragments de DNA de tailles connues pour servir de marqueurs de taille .")
87
On ajoute dans les dépôts un colorant bien visible sur le gel qui va migrer très rapidement avant les fragments de DNA pour limiter la distance parcourue au bord de l’anode . On examine le gel sous lumière ultra-violette à 254 nm et on photographie les fragments d’ADN digéré après coloration de l’ADN par le bromure d’ éthidium ( Bet)
")
92
Chromatographie des AN
Les méthodes Chromatographiques sont plus utilisées pour purifier que pour séparer

93
La chromatographie d’adsorption sur colonne de silice ; purification
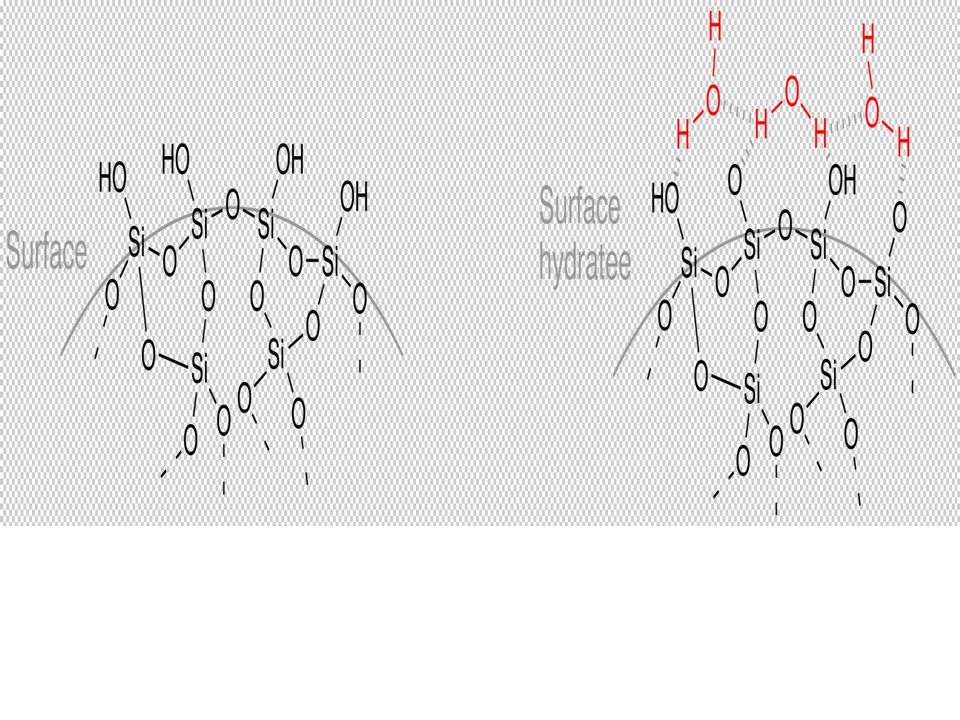
Cette technique est actuellement largement utilisée et des kits d’extraction basés sur ce principe sont proposés par de nombreux fournisseurs, adaptés à de nombreux échantillons. Le support Solide /silice SiO2 :

95
- Les interactions entre les AN et la silice (chromatographie par adsorption) La fixation de l’ADN sur la silice fait intervenir des interactions dipôle-dipôle

96
Les étapes de la chromatographie
en présence de concentrations élevées en sel [force ionique élevée([NaCl]] et en présence d’alcool : L’agent chaotropique intervient comme compétiteur dans les relations que l’ADN établit avec l’eau.

97
Le sel se lie à l’eau et l’ADN devient moins hydraté
Le sel se lie à l’eau et l’ADN devient moins hydraté. L’ADN établit alors des liaisons avec la silice et se fixe au support de la colonne. Les autres macromolécules restent en solution La colonne est « lavée » par de l’éthanol, solvant moins polaire que l’eau qui ne risque pas de décrocher l’ADN. L’ADN est élué par un tampon à basse force ionique. Méthode rapide

98
Purification des ARNm Les ARNm se caractérisent par la présence du côté 3’-terminal d’une longue queue poly(A) synthétisée à la phase post-transcriptionnelle. On utilise une chromatographie d’affinité entre cette queue poly(A) et une colonne dont la phase fixe est pourvue de fragments d’oligo(dT) de quelques dizaines de nucléotides qui peuvent s’hybrider avec les ARN poly(A).Les interaction sont donc du type A=T
 synthétisée à la phase post-transcriptionnelle. On utilise une chromatographie d’affinité entre cette queue poly(A) et une colonne dont la phase fixe est pourvue de fragments d’oligo(dT) de quelques dizaines de nucléotides qui peuvent s’hybrider avec les ARN poly(A).Les interaction sont donc du type A=T.")
99
Protocole Lavage de la colonne en milieu alcalin (potasse),
Dépôt des RNA totaux à haute force ionique (KCl 0,5 M) Rincage de la colonne avec du KCl 0,1 M en surveillant l’absorbance de l’éluat à 260 nm pour s’assurer de l’élimination des RNA non retenus par la colonne (rRNA, tRNA,... ). Elution de la fraction retenue par la colonne en abaissant la concentration de KCl à 0,01 M et en élevant la température. Un micro essai avec la reverse transcriptase permet de s’assurer de la qualité de ce RNA poly(A) comme matrice pour la synthèse de cDNA
 Rincage de la colonne avec du KCl 0,1 M en surveillant l’absorbance de l’éluat à 260 nm pour s’assurer de l’élimination des RNA non retenus par la colonne (rRNA, tRNA,... ). Elution de la fraction retenue par la colonne en abaissant la concentration de KCl à 0,01 M et en élevant la température. Un micro essai avec la reverse transcriptase permet de s’assurer de la qualité de ce RNA poly(A) comme matrice pour la synthèse de cDNA.")
100
Dosage des AN Les bases puriques et pyrimidiques absorbent à 260nm ( UV) 1 unité de densité optique à 260nm correspond : - pour une solution d'ADN double brin à 50µg/ml - pour une solution d'ARN ou d'ADN simple brin à 25µg/ml Ces valeurs s'appliquent à des acides nucléiques parfaitement purs et en solution homogène.

101
Contrôle de pureté rechercher une éventuelle contamination protéique car les protéines absorbent non seulement à 280nm mais aussi à 260nm. Pour cette raison on effectue une seconde mesure de DO à 280nm. Un ADN pur doit avoir un rapport DO 260 / DO 280 compris entre 1,8 et 2.Une éventuelle contamination par du phénol peut être recherchée en mesurant l'absorption à 270nm.

102
Calcul Exemple : [ ADN] = DO260 X 50µg/ml X facteur de dilution [ ADN] = 0,256 X 50µg/ml X 20 ( 5µl d'ADN dans 95µl d'eau, dilution au 20eme)= 152µg/ml ou 152ng/µl Quantité totale = 152 X 50µl = 7600ng ou 7,6µg
= 152µg/ml ou 152ng/µl Quantité totale = 152 X 50µl = 7600ng ou 7,6µg.")
103
Méthodes d’Identification d’un Fragment d’ADN
Gène ou partie d’un gène dans un but diagnostic

104
Le Southern Blot E. Southern ( Prof Biochimie Oxford, UK)
Exemples Traités au tableau Animation vidéo sur le web

105
1-Le gel d'électrophorèse est trempé dans une solution alcaline (contenant typiquement de l'hydroxyde de sodium) afin de permettre la dénaturation de l'ADN bicaténaire (séparation de l'ADN double-brin en simple-brin).
 afin de permettre la dénaturation de l ADN bicaténaire (séparation de l ADN double-brin en simple-brin).")
106
2-Une feuille de membrane de nylon est placée sur le gel
2-Une feuille de membrane de nylon est placée sur le gel. Une pression est appliquée au gel en plaçant une pile de papier absorbant et par un poids sur le dispositif . Ceci va permettre le « buvardage ( blot) » de l'ADN contenu dans le gel sur la membrane, où il va se fixer ( par capillarité)
 » de l ADN contenu dans le gel sur la membrane, où il va se fixer ( par capillarité)")
107
3-La membrane est exposée au rayonnement ultra-violet afin de fixer de manière permanente l'ADN sur le nylon

108
4-La membrane est ensuite mise en contact avec une sonde spécifique de la séquence d'ADN recherchée. La sonde est marquée de sorte qu'elle puisse être détectée, habituellement en incorporant de la radioactivité ou en "étiquetant" la molécule avec un fluorophore. Dans certains cas, la sonde peut être faite à partir d'ARN, plutôt que d'ADN.

109
5-Après hybridation, la sonde en excès est éliminée de la membrane par différents lavages, et l'hybridation est visualisée sur un film autoradiographique, dans le cas d'une sonde radioactive ou fluorescente.

110
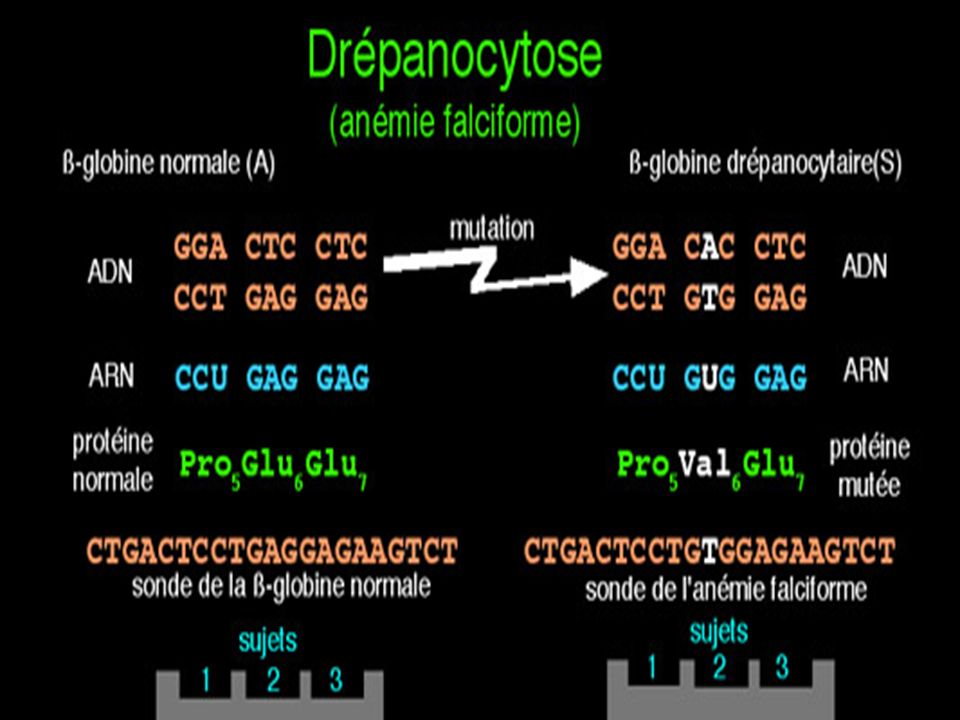
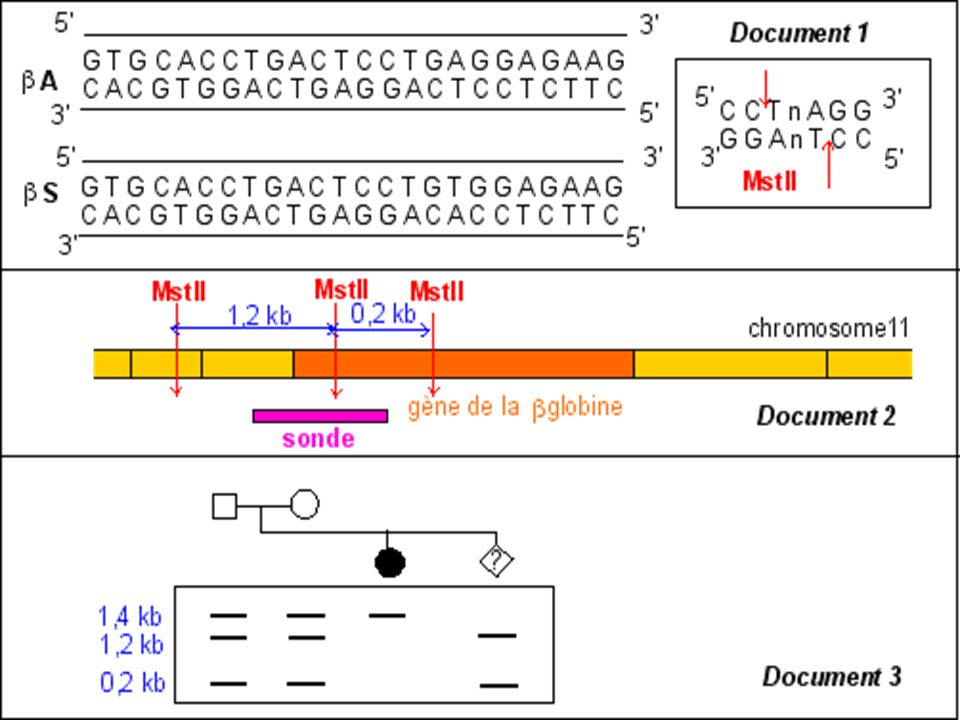
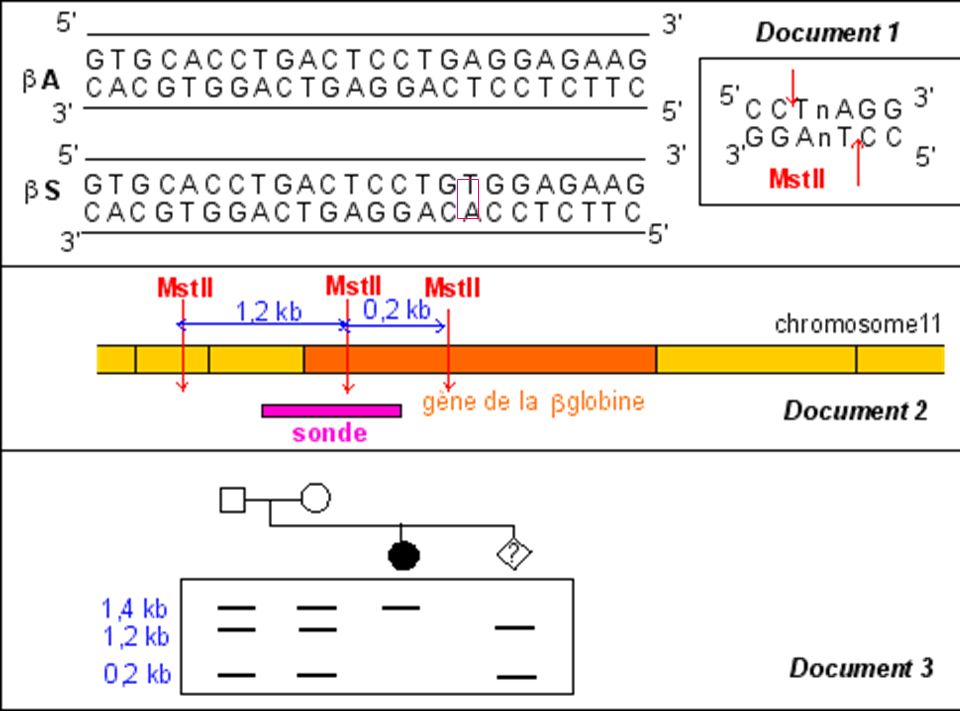
Diagnostic moléculaire du gène de la Drépanocytose par S. Blot
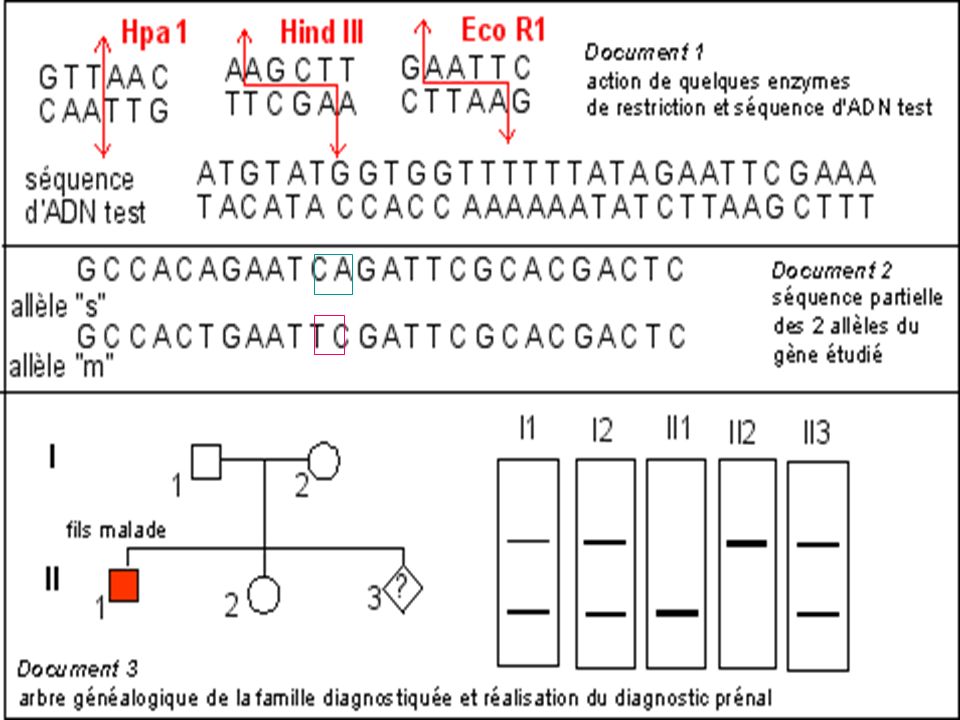
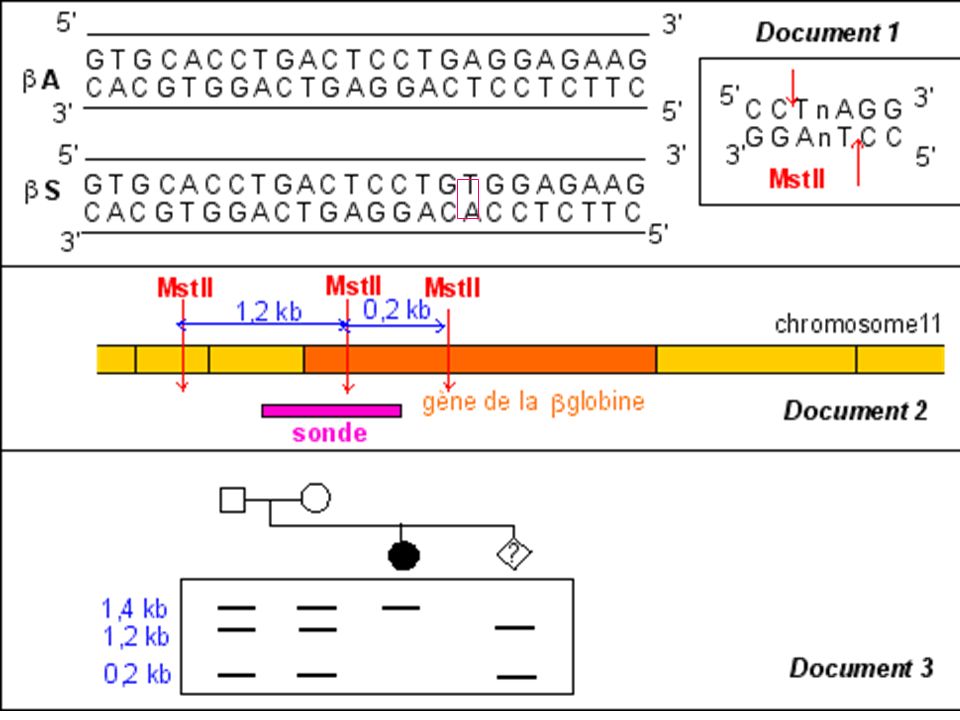
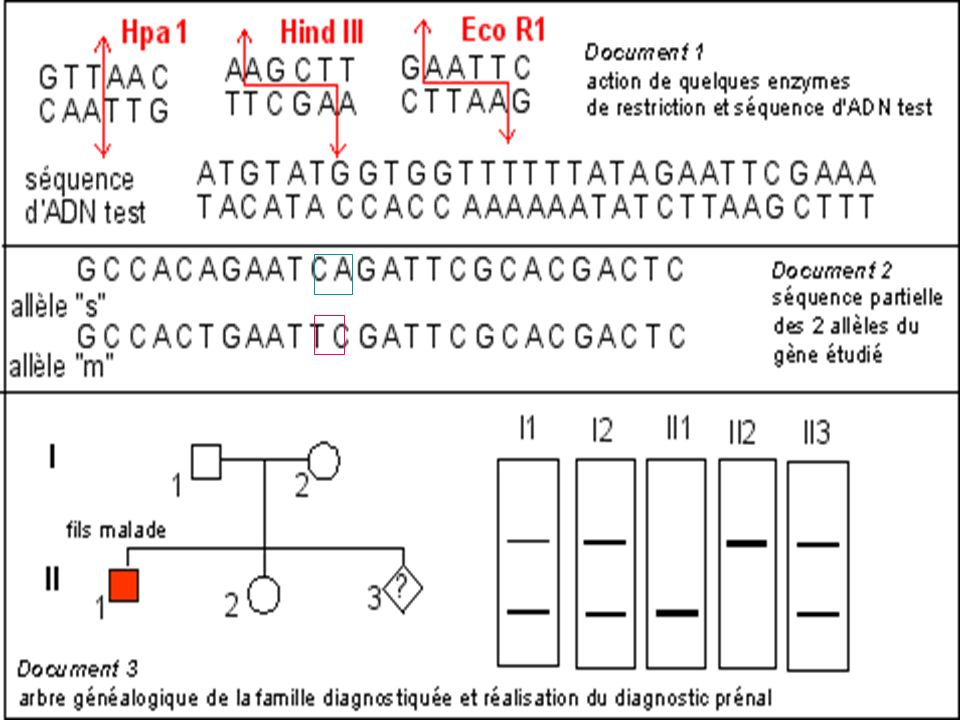
La mutation qui conduit à l'allèle muté b S supprime un site de restriction ( MstII) Avec une sonde appropriée le fragment d'ADN muté digéré par MstII génère (après réalisation de la méthode de Southern) un fragment de 1.4 kb alors que 2 fragments (1.2 kb kb) correspondent au(x) gène(s) sauvage(s).
 Avec une sonde appropriée le fragment d ADN muté digéré par MstII génère (après réalisation de la méthode de Southern) un fragment de 1.4 kb alors que 2 fragments (1.2 kb kb) correspondent au(x) gène(s) sauvage(s).")
113
LES Méthodes PCR

114
III- LES APPLICATIONS de la PCR
Polymerase chain reaction

115
Avantage de la Technique
Elle permet d’étudier un segment d’ ADN au sein de tout le génome si l’on connaît sa séquence ou une partie de sa séquence Elle permet de recopier ce segment (amplification des millions de fois et de le visualiser par une méthode électrophorétique Par coloration au bromure d’éthidium ou autre colorant Ce segment peut être par la suite traité pour mettre en évidence une mutation ( RFLP) Pour le cloner
 Pour le cloner.")
116
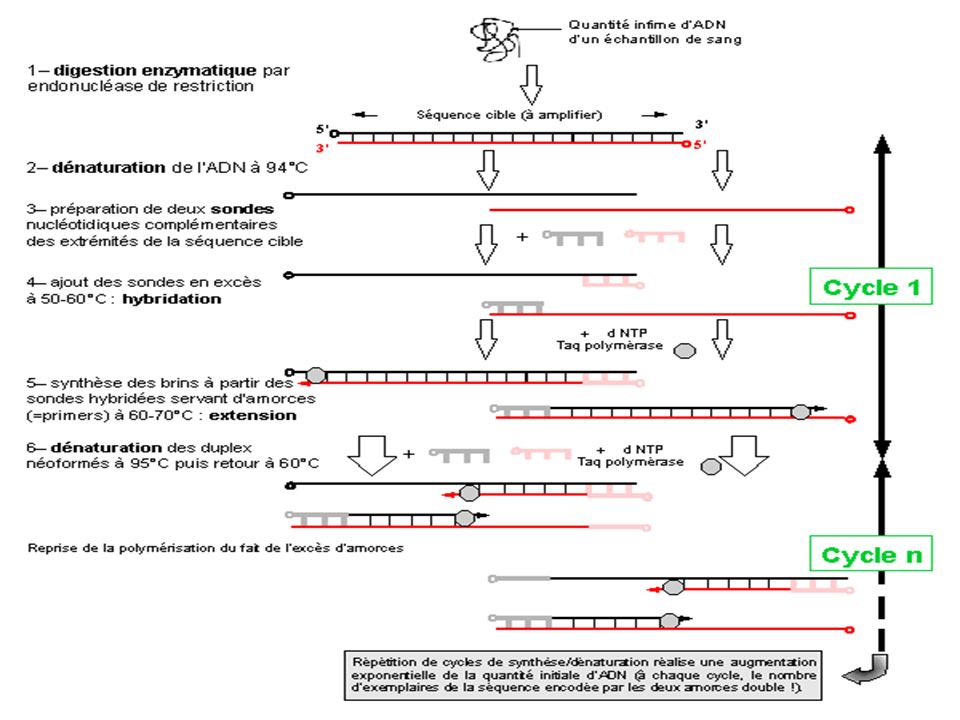
Principe Succession de réactions de réplication d'une matrice double brin d'ADN. Chaque réaction met en oeuvre deux amorces oligonucléotidiques dont les extrémités 3-prime pointent l'une vers l'autre. Les amorces ou «primers» en anglais définissent alors, en la bornant, la séquence à amplifier. Les amorces sont orientées dans le sens 5’ vers 3’

117
Chaque produit de chaque étape de synthèse sert de matrices pour les étapes suivantes. Au lieu d'être linéaire, l'amplification obtenue exponentielle. Imaginée par K. Mullis en 1985 (Prix Nobel de Chimie dès 1993), la technique connaît un essor considérable à partir de la commercialisation (vers 1988), d'une ADN polymérase résistante aux températures élevées (la Taq polymérase)
, la technique connaît un essor considérable à partir de la commercialisation (vers 1988), d une ADN polymérase résistante aux températures élevées (la Taq polymérase)")
118
On opère avec des sauts de température à chaque étape
EN effet, une température dite d’annealing permet l’hybridation de l’amorce avec l’ADN ( 56°) A une autre température , c’est l’ élongation ou extension ( 72 ° C) A une dernière température les brins amplifiés sont séparés : température de dénaturation ~~ 94 °C
 A une autre température , c’est l’ élongation ou extension ( 72 ° C) A une dernière température les brins amplifiés sont séparés : température de dénaturation ~~ 94 °C.")
120
Ou Principe de la PCR > ( google)

121
Les longueurs d’ADN que l’on peut amplifier sont couramment d’environ 800-1000 pb
Des amplifications plus longues sont possibles mais requièrent des Taq particulières Des difficultés peuvent survenir lorsque les régions sont très riches en GC

123
B- GAP-PCR / Application de la méthode PCR au diagnostic de grandes délétions
Des amorces choisies pour être très éloignées l’une de l’autre, ne permettront pas un amplification par une Taq pol classique En cas de délétion, les amorces sont rapprochées et l’ADN amplifié signe alors une délétion.

124
Distance entre A4 et A9 = 1800 pb : si les gènes alpha 1 et alpha 2 sont présents la
Région amplifiée est de 194pb entre A4 et A1B. Si ,les deux gènes sont délétés la distance entre A4 et A9 est raccourcie et l’amplification révèle un fragment de 550pb.

125
C- LA PCR INVERSE ou Reverse Transcriptase PCR ( RT-PCR)
La RT-PCR a été mise au point pour utiliser les ARN comme matrice d'amplification de la PCR On isole des ARN m par purification spécifique de ces derniers Ils sont ensuite « recopiés » en ADN par une enzyme : la transcriptase reverse On réalise une PCR comme précédemment

126
Le produit final est un ADN dont l'un des brins est complémentaire de l'ARN d'intérêt et l'autre brin a la même séquence que cet ARN d'intérêt (à la substitution près de U par T).
.")
128
Difficultés Techniques
La contamination des échantillons par de l'ADN génomique est une des principales difficulté de cette technique. En effet, l'ADN génomique peut entrer en compétition avec l'ADNc pour lors de l'étape d'hybridation des amorces. Diverses parades sont alors utilisées :

129
Parades 1 Toujours purifier les ARNm sur une colonne où sont immobilisés des oligo-dT

130
2 Les ADN présents dans l'échantillon peuvent être détruits par ajout d'une activité désoxyribonucléase (dépourvue d'activité ribonucléase).
.")
131
3 Choisir les amorces aux bornes d'un intron
3 Choisir les amorces aux bornes d'un intron. Ainsi, les produits d'amplification issus de l'ADNc et ceux issus d'ADN génomique contaminant seront distingués selon leur taille : les fragments issus de l'ADN génomique (contenant l'intron) auront une taille supérieure aux fragments d'amplification issus de l'ADNc (ne contenant pas l'intron).
 auront une taille supérieure aux fragments d amplification issus de l ADNc (ne contenant pas l intron).")
132
D- MUTAGENESE DIRIGEE Réalisation d’une amorce mutée
l'extrémité 3' doit rester complémentaire de la séquence de matrice à amplifier (initiation de la polymérisation) l'extrémité 5' qui peut porter de nombreuses modifications : des mutations ponctuelles, des sites de restriction, des séquences de recombinaison etc.
 l extrémité 5 qui peut porter de nombreuses modifications : des mutations ponctuelles, des sites de restriction, des séquences de recombinaison etc.")
134
Mutation à des extrémités

136
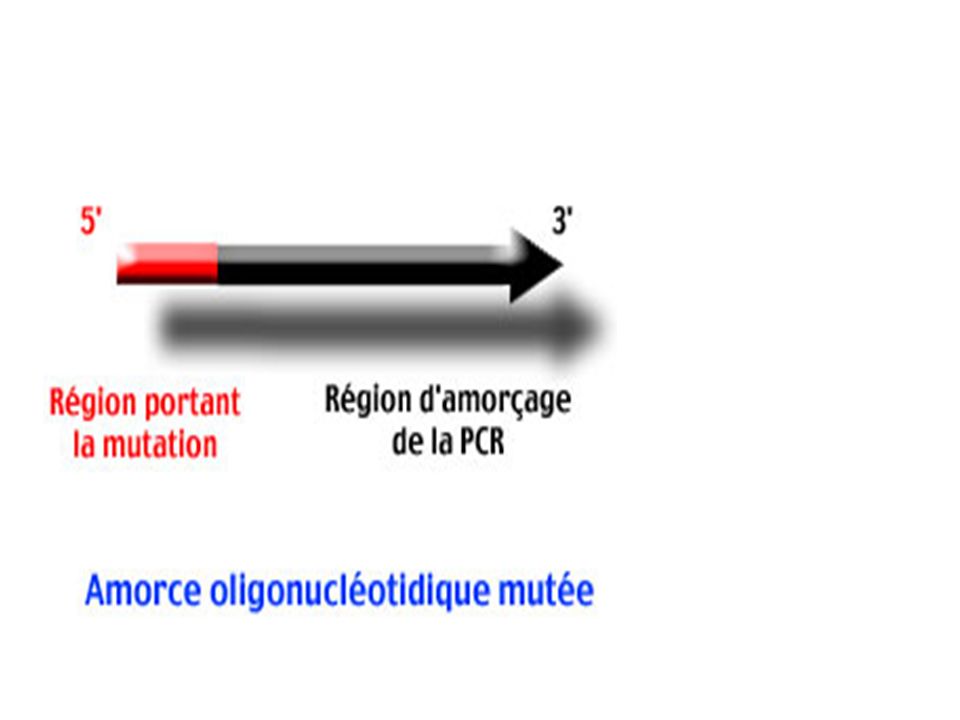
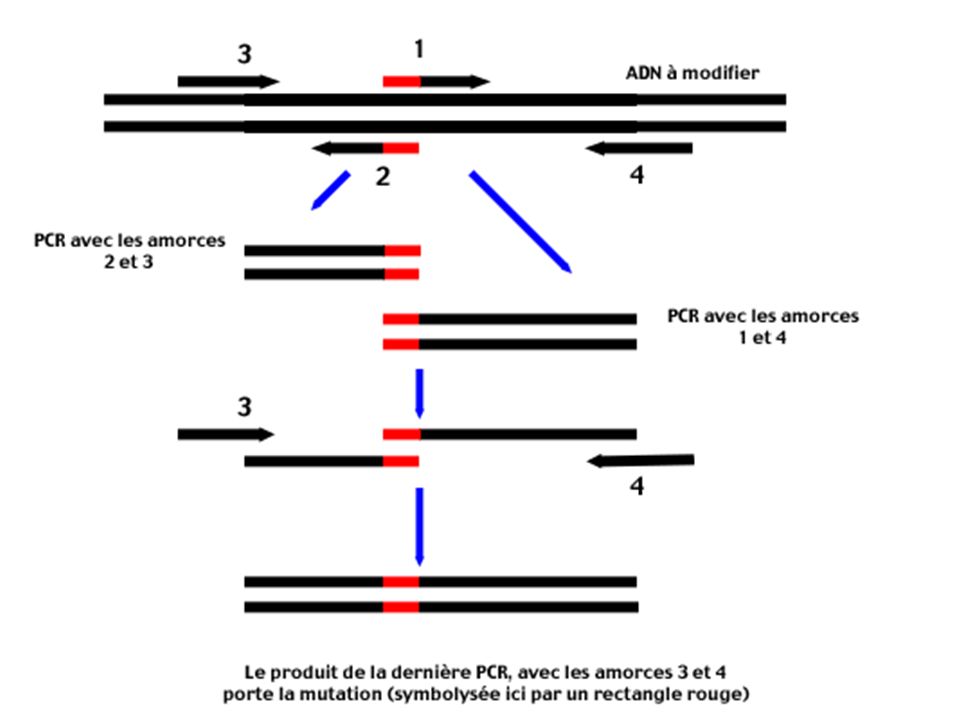
Principe Les amorces 1 et 2 sont des amorces mutées (la région 5' qui porte la mutation est symbolisée par un rectangle rouge). Obtenues par synthèse chimique . Ces amorces sont complémentaires (le recouvrement des régions 5' mutées en rouge est particulièrement important pour la suite). Les amorces 3 et 4 bornent le segment d'ADN à modifier. Elles détermineront la longueur du produit final.
. Obtenues par synthèse chimique . Ces amorces sont complémentaires (le recouvrement des régions 5 mutées en rouge est particulièrement important pour la suite). Les amorces 3 et 4 bornent le segment d ADN à modifier. Elles détermineront la longueur du produit final.")
137
Deux réactions de PCR sont effectuées en parallèle : Une PCR avec les amorces 2 et 3 donne un produit d'amplification correctement muté dans la région désirée, mais de petite taille

139
Une PCR avec les amorces 1 et 4 donne aussi un produit muté, mais toujours de taille insuffisante.

140
Les deux produits de PCR sont purifiés , il s'agit surtout d'éliminer les amorces 1 et 2
On regroupe dans le même tube tous les produits PCR

141
Ils possèdent en commun la région mutée (région d'ADN possédant un rectangle rouge). L'hybridation de ces deux fragments par cette partie commune est critique pour la dernière étape.

142
Une dernière PCR utilisant les amorces 3 et 4 est alors réalisée sur ce mélange et donne, en effet, un fragment de la taille souhaitée contenant la mutation.

144
IV LE SEQUENCAGE ADN ARN

145
Le séquençage de l’ADN

146
Chaque espèce d’ARN va hybrider avec la sonde d’ADN correspondante sur la puce
préalablment chargée de sondes d’ADN. La combinaison ARN biotinylé – ADN est fluorescente et donc repérable par un système de lecture approprié Fluorescent Stain Biotin RNA After staining, RNA (purple) bound to the DNA probe built on the array will fluoresce RNA (purple) has bound to its DNA probe built on the array
 bound to the DNA probe built on the array will fluoresce. RNA (purple) has bound to its DNA probe built on the array.")
147
Séquençage de l’ADN . F SANGER (1977)
")
148
Séquençage de l’ADN :Walter GILBERT

149
LE SEQUENCAGE PAR LA METHODE DE SANGER
On copie le brin à séquencer de telle sorte que les copies soient radioactives (ou repérables par un autre marquage : fluorescence ) Soit la séquence suivante à déterminer : 3’ …… G C A T G A T C G G 5’ ……Amorce présentera une extrémité 3’ OH libre Il faut choisir une amorce complémentaire d’un bout de séquence connue
 Soit la séquence suivante à déterminer : 3’ …… G C A T G A T C G G 5’ ……Amorce présentera une extrémité 3’ OH libre. Il faut choisir une amorce complémentaire d’un bout de séquence connue.")
151
La DNA polymérase réplique 5' 3' à partir 3'OH libre de l’amorce
en présence d'un 2, 3 didéoxynucléotide la réplication est stoppée. Statistiquement on observera des fragments d'ADN de tailles différentes en fonction de l'incorporation du 2, 3 didéoxynucléotide pendant la réplication du brin à séquencer

152
Tube 1 4dNTP, ddGTP 3' GCATGATCGG CG# CGTACTAG# Tube 2 4dNTP, ddATP 3'GCATGATCGG CGTA# CGTACTA#

153
Tube 3 4dNTP, ddCTP 3' GCATGATCGG 5' ddC # CGTAC# CGTACTAGC# CGTACTAGCC# Tube 4 4dNTP, ddTTP CGT# CGTACT#

154
Tube 1 : ddGTP 1er fragment : 2 bases 2nd fragment : 8 bases Tube 2 : ddATP 1er fragment : 4 bases 2nd fragment : 7 bases Tube 3 : ddCTP 1er fragment : 1 base 2nd fragment : 5 bases 3ème fragment : 9 bases 4ème fragment : 10 bases Tube 4 : ddTTP 1er fragment : 3 bases 2nd fragment : 6 bases

155
Remarques : Il n'y a jamais deux fois la même taille quelque soit le tube examiné Les produits sont radioactifs, ils vont être repérés par autoradiographie après électrophorèse en gel d'acrylamide. Les fragments migreront d'autant plus vite que leur masse moléculaire est faible. La lecture se fera de 5’ vers 3 ‘ en commençant par les fragments les plus petits ( du bas vers le haut)
")
156
On lit donc Lecture : 5' CGTACTAGCC 3' Séquence : 3' GCATGATCGG 5‘ On donne le produit complémentaire du produit de lecture : 3’ …… G C A T G A T C G G 5’

158
Sanger Centre-Cambridge

159
SEQUENCAGE DIRECT DE l‘ARN
Les Principes Avec transformation en c DNA Sans tranformation en c DNA

160
Transformation en cDNA
UN principe ( parmis d’autres …) l
 l.")
161
Fragmentation des ARN messagers.
(b) Synthèse du premier brin de cDNA avec une amorce hexamère aléatoire marqué par une séquence flanquante (FDV). (c) Des cytosines sont ajoutées à chaque cDNA (d) Une hybridation entre le cDNA (vert) et un fragment d'ARN marqué par une seconde séquence flanquante (RDV) permet d'incorporer un site de marquage RDV dans le premier brin de cDNA. (e) La banque est amplifiée par PCR avec des amorces FDV et RDV. (f) Les fragments amplifiés sont attachés à des billes par émulsion. (g) Les billes sont fixées à un support.
 Synthèse du premier brin de cDNA avec une amorce hexamère aléatoire marqué par une séquence flanquante (FDV). (c) Des cytosines sont ajoutées à chaque cDNA. (d) Une hybridation entre le cDNA (vert) et un fragment d ARN marqué par une seconde séquence flanquante (RDV) permet. d incorporer un site de marquage RDV dans le premier brin de cDNA. (e) La banque est amplifiée par PCR avec des amorces FDV et RDV. (f) Les fragments amplifiés sont attachés à des billes par émulsion. (g) Les billes sont fixées à un support.")
162
Sans transformation en cDNA
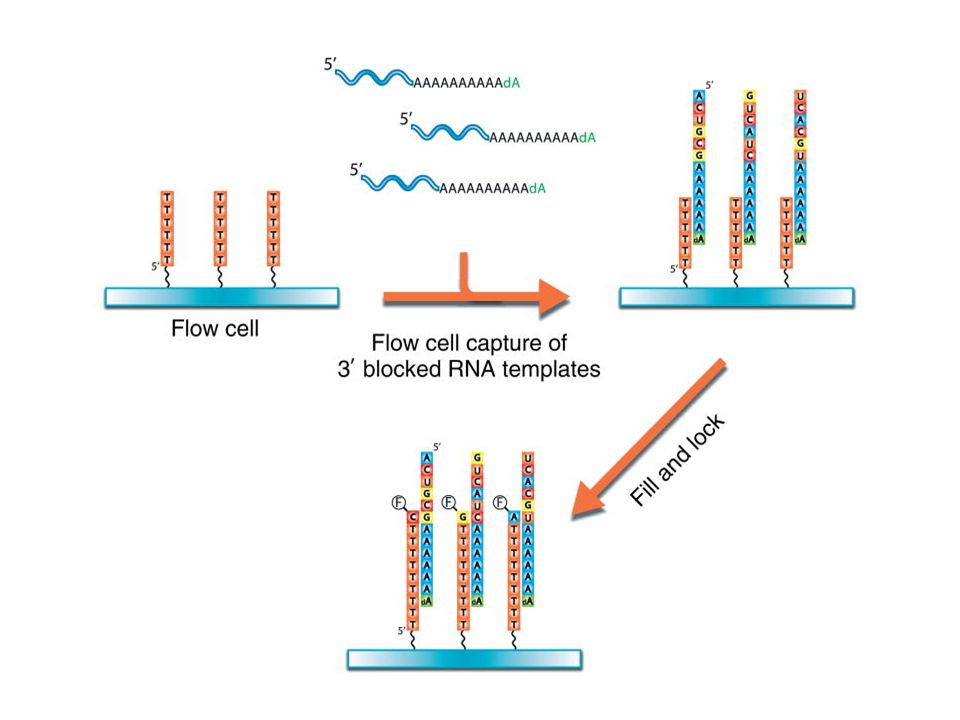
Purication des ARNm ( oligo dT) Des séquences poly T sont bloquées sur un support Les ARNs sont bloqués en 3’ par une déoxy thymidine Le support capture les ARN par leur extrémité poly A A chaque nucléotide incorporé correspond un signal fluorecent témoin du nucléotide incorporé qui permet de lire une séquence
 Des séquences poly T sont bloquées sur un support. Les ARNs sont bloqués en 3’ par une déoxy thymidine. Le support capture les ARN par leur extrémité poly A. A chaque nucléotide incorporé correspond un signal fluorecent témoin du nucléotide incorporé qui permet de lire une séquence.")
163
Sequençage direct de l’ARN
ON ne transforme pas en cDNA Des séquences de poly T sont bloquées sur un support Le support capture les ARNm purifiés par leur extrémité poly A 3’dA : un 3’ di Desoxy A bloque la fin de séquence d’ARN ( ne permettra pas la réplication vers le bas) La séquence débute par allongement du brin DNA avec l’ARNm qui sert de matrice Les ARN sont ensuite complétés avec addition de Thymidine non marquée afin de ne pas laisser le dernier A libre sur l’ARN)
 La séquence débute par allongement du brin DNA avec l’ARNm qui sert de matrice. Les ARN sont ensuite complétés avec addition de Thymidine non marquée afin de ne pas laisser le dernier A libre sur l’ARN)")
164
IL faut disposer d’une Polymérase spécifique pour répliquer à partir d’une matrice ARN
La séquence débute par réplication du premier nucléotide en 5’ du dernier A . Le nucléotide incorporé est fluorescent (VF dit terminateurs) il ne permet pas l’addition du nucléotide suivant sans élimination du composé VF fluorescent. Le VF est libéré par traitement chimique et déclenche un signal fluorescent témoin du nucléotide incorporé Au nucléotide libéré du VF va s’ajouter le nucléotide suivant et la réplication-séquence va pouvoir être poursuivie comme précédement Cette méthode permet le début d’une lecture par fluorescence là où commence le RNA et ne lit pas le poly A .
 il ne permet pas l’addition du nucléotide suivant sans élimination du composé VF fluorescent. Le VF est libéré par traitement chimique et déclenche un signal fluorescent témoin du nucléotide incorporé. Au nucléotide libéré du VF va s’ajouter le nucléotide suivant et la réplication-séquence va pouvoir être poursuivie comme précédement. Cette méthode permet le début d’une lecture par fluorescence là où commence le RNA et ne lit pas le poly A .")
167
Etude des Interactions ADN-Protéines
De nombreux facteurs sont requis pour la régulation de l’expression des gènes dans le temps et dans l’espace de façon précise.

168
I-Sensibilité à la DNAse I
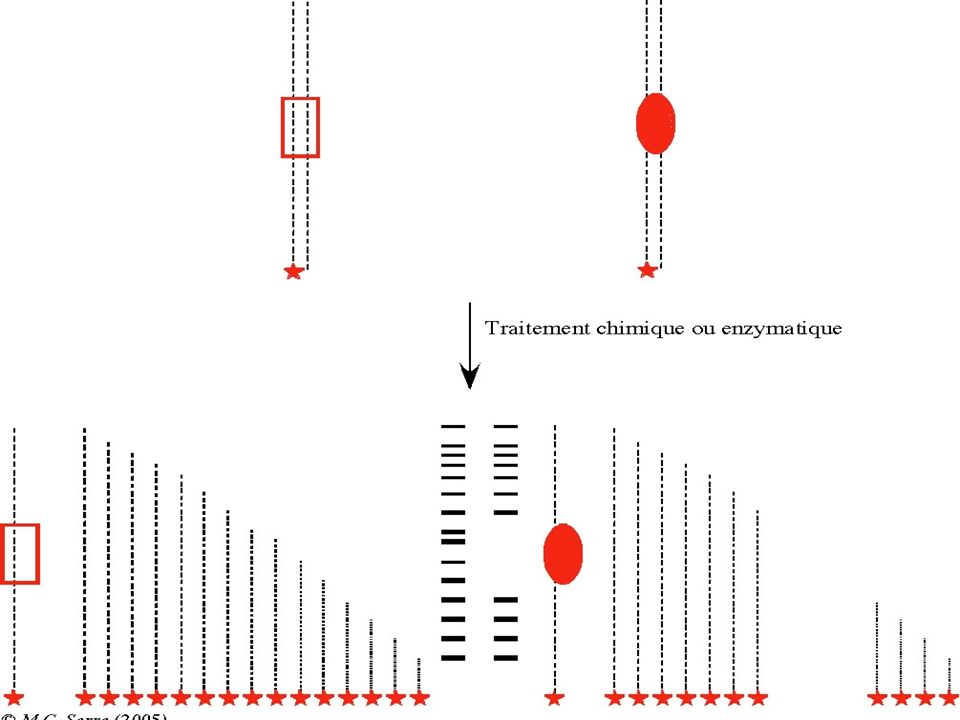
BUT : Révéler la fixation d’une protéine à l’ADN pour identifier des sites d’interaction ADN-Protéine (Transcription & Régulation ) Principe : La fixation d’une protéine à un fragment d’ ADN réduit sa mobilité électrophorètique . La fixation d’une protéine à l’ADN protège ce dernier contre une dégradation par la DNAse I. Technique : Foot Printing
 Principe : La fixation d’une protéine à un fragment d’ ADN réduit sa mobilité électrophorètique . La fixation d’une protéine à l’ADN protège ce dernier contre une dégradation par la DNAse I. Technique : Foot Printing.")
169
Pour un ADN marqué à une seule extrémité, l’endroit d’un clivage par une DNAse sera déduit à partir de la taille du fragment marqué. La taille sera déterminée par électrophorèse après repérage du fragment marqué. Si une protéine recouvre une séquence d’ ADN, elle « protège » l’ADN contre l’action de la DNAse. L’empreinte de la portion recouverte par une protéine sera révélée par une absence de bandes sur le gel.

171
Augmentation [P] >>>
![Augmentation [P] >>>](http://slideplayer.fr/slide/9480482/29/images/171/Augmentation+%5BP%5D+%3E%3E%3E.jpg "Augmentation [P] >>>")
172
II-Immuno-Précipitation de la Chromatine
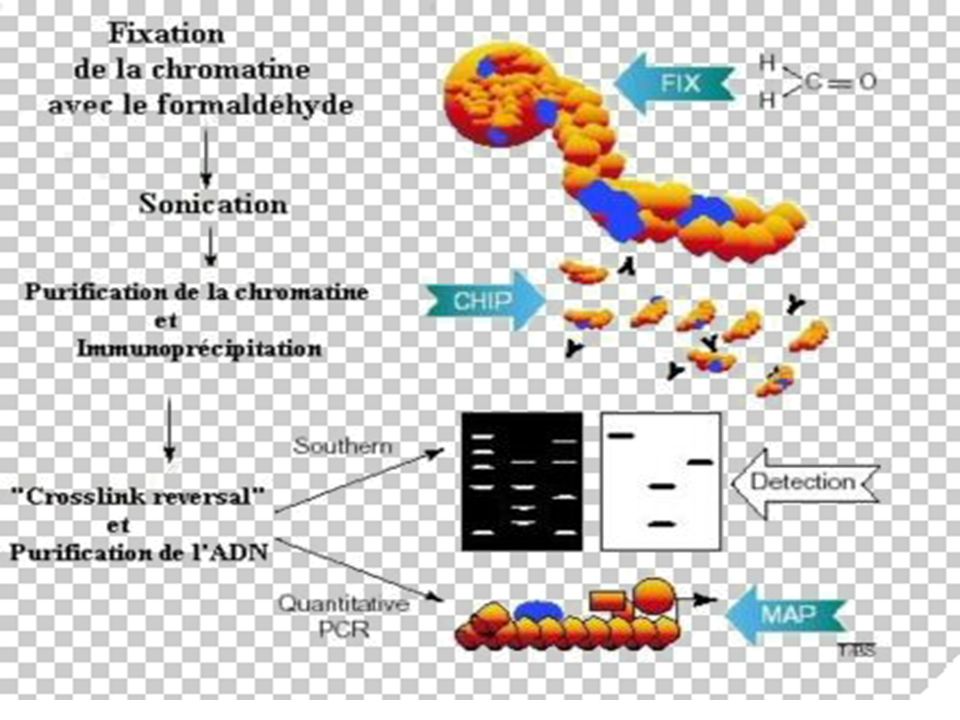
BUT La méthode ChIP (ImmunoPrécipitation de la Chromatine) permet [sur des cellules entières] d’identifier des protéines associées à une région spécifique du génome et inversement, d’identifier différentes régions du génome associées à une protéine particulière. Le principe de cette technique est basé sur la fixation des protéines à l’ADN par le formaldéhyde, suivie d’une immuno-précipitation par un AC spécifique
 permet [sur des cellules entières] d’identifier des protéines associées à une région spécifique du génome et inversement, d’identifier différentes régions du génome associées à une protéine particulière. Le principe de cette technique est basé sur la fixation des protéines à l’ADN par le formaldéhyde, suivie d’une immuno-précipitation par un AC spécifique.")
173
Principe de la Technique
Les liaisons protéines-ADN sont renforcées par traitement au formaldéhyde. L'ADN est ensuite clivé en fragments de 500 à 1000 pb par sonication. La protéine d'intérêt est immuno-précipitée, l'ADN sur lequel elle était liée est récupéré et amplifié par PCR avec des amorces choisies sur les régions promotrices d'intérêt.

174
Avantages : Cette technique est utilisée sur des cellules vivantes (plus précis qu'un essai "in vitro" par gel shift par exemple) Elle permet non seulement de visualiser l'occupation des sites sur les promoteurs à un moment voulu, en réponse à des traitements spécifiques, mais aussi les modifications post-traductionnelles des facteurs de transcription d'intérêt suivant les anticorps utilisés. L'utilisation d'anticorps anti-histones acétylés permet de visualiser l'état général de la chromatine au niveau de promoteurs spécifiques.

175
Grâce à cette méthode, il est possible de récupérer et de purifier des protéines qui interagissent avec des récepteurs, mais aussi des séquences génomiques adjacentes aux facteurs de transcriptions. Après la purification de ces complexes, les séquences nucléotidiques peuvent être clonées, séquencées et analysées.

177
Cartographie de l’expression des GENES
Hybridation des ARNm sur puces à ADN

178
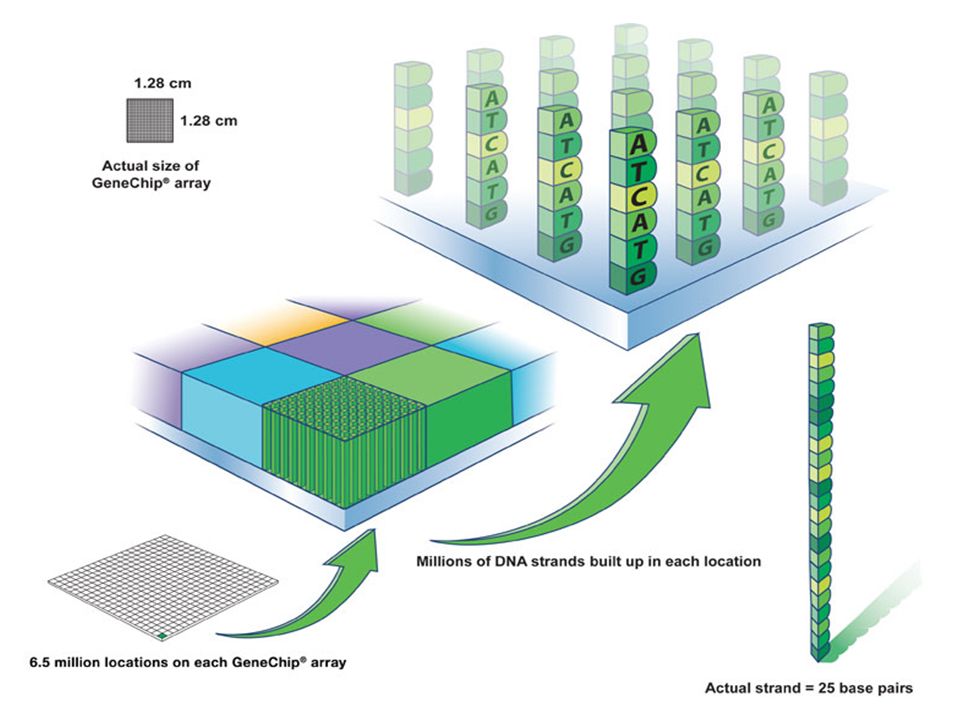
Puce à ADN

180
Analyse totale du génome par utilisation de
l’expression différentielle des gènes On construit des supports ou chip-arrays . Chaque grille est un carré de 11 micromètres porteur de séquences d’ ADN (oligoprobe ( complémentaire à celle de séquences de gènes) Des Millions de brins de DNA gréffés et accessibles 6.5 millions de brin sur chaque chip Brins = 25 base pairs
 Des Millions de brins de DNA gréffés et accessibles. 6.5 millions de brin sur chaque chip. Brins = 25 base pairs.")
181
Isoler L’ARN de cellules d’intérêt
Analyse du génome Isoler L’ARN de cellules d’intérêt Marquage des ARNs à la biotine et hybridation par le système des microarray Biotin-labelled RNA Biotin label the RNA and hybridise to the microarray

182
Chaque espèce d’ARN va hybrider avec la sonde d’ADN correspondante sur la puce
préalablment chargée de sondes d’ADN. La combinaison ARN biotinylé – ADN est fluorescente et donc repérable par un système de lecture approprié Fluorescent Stain Biotin RNA After staining, RNA (purple) bound to the DNA probe built on the array will fluoresce RNA (purple) has bound to its DNA probe built on the array
 bound to the DNA probe built on the array will fluoresce. RNA (purple) has bound to its DNA probe built on the array.")
183
Chaque espèce d’ARN va hybrider avec la sonde d’ADN correspondante sur la puce
préalablment chargée de sondes d’ADN. La combinaison ARN biotinylé – ADN est fluorescente et donc repérable par un système de lecture approprié Fluorescent Stain Biotin RNA After staining, RNA (purple) bound to the DNA probe built on the array will fluoresce RNA (purple) has bound to its DNA probe built on the array
 bound to the DNA probe built on the array will fluoresce. RNA (purple) has bound to its DNA probe built on the array.")
184
Résultats L’intensité du signal reflète l’abondance des ARNs qui se sont hybridés à l’AND de la puce Red =expression importante Vert = intermédiare Noir = pas d’expression

185
Echantillon 1 Echantillon 2 Comparaison de l’expression d’ARN avant (1) et après traitement (2) anti tumoral . Il est possible de grouper des expressions de gènes Sans connaître leur fonction

186
ETUDE DE L’EXPRESSION DES GENES

187
Etude des Interactions ADN-Protéines
De nombreux facteurs sont requis pour la régulation de l’expression des gènes dans le temps et dans l’espace de façon précise.

188
I-Sensibilité à la DNAse I
BUT : Révéler la fixation d’une protéine à l’ADN pour identifier des sites d’interaction ADN-Protéine (Transcription & Régulation ) Principe : La fixation d’une protéine à un fragment d’ ADN réduit sa mobilité électrophorètique . La fixation d’une protéine à l’ADN protège ce dernier contre une dégradation par la DNAse I. Technique : Foot Printing
 Principe : La fixation d’une protéine à un fragment d’ ADN réduit sa mobilité électrophorètique . La fixation d’une protéine à l’ADN protège ce dernier contre une dégradation par la DNAse I. Technique : Foot Printing.")
189
Questions posées Pourquoi après dénaturation de l’ ADN dans les chromosomes ( FISH) ceux-ci sont –ils chromosomes encore visibles ? Fixation de la Taq polymérase

190
Pour un ADN marqué à une seule extrémité, l’endroit d’un clivage par une DNAse sera déduit à partir de la taille du fragment marqué. La taille sera déterminée par électrophorèse après repérage du fragment marqué. Si une protéine recouvre une séquence d’ ADN, elle « protège » l’ADN contre l’action de la DNAse. L’empreinte de la portion recouverte par une protéine sera révélée par une absence de bandes sur le gel.

192
Augmentation [P] >>>
![Augmentation [P] >>>](http://slideplayer.fr/slide/9480482/29/images/192/Augmentation+%5BP%5D+%3E%3E%3E.jpg "Augmentation [P] >>>")
193
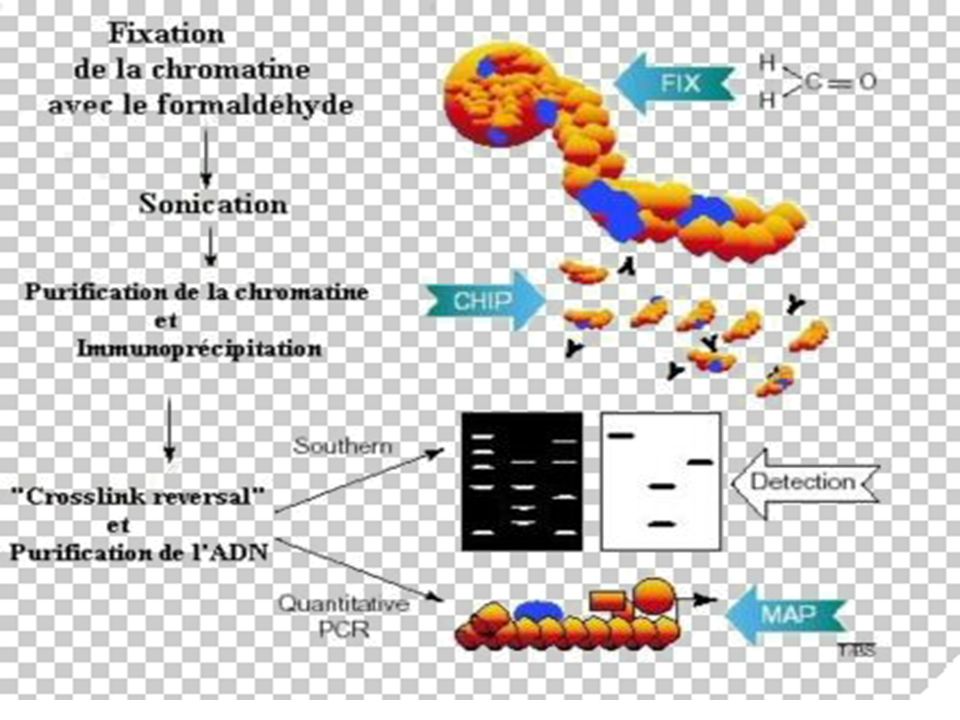
II-Immuno-Précipitation e la Chromatine
BUT La méthode ChIP (ImmunoPrécipitation de la Chromatine) permet [sur des cellules entières] d’identifier des protéines associées à une région spécifique du génome et inversement, d’identifier différentes régions du génome associées à une protéine particulière. Le principe de cette technique est basé sur la fixation des protéines à l’ADN par le formaldéhyde, suivie d’une immuno-précipitation par un AC spécifique
 permet [sur des cellules entières] d’identifier des protéines associées à une région spécifique du génome et inversement, d’identifier différentes régions du génome associées à une protéine particulière. Le principe de cette technique est basé sur la fixation des protéines à l’ADN par le formaldéhyde, suivie d’une immuno-précipitation par un AC spécifique.")
194
Principe de la Technique
Les liaisons protéines-ADN sont renforcées par traitement au formaldéhyde. L'ADN est ensuite clivé en fragments de 500 à 1000 pb par sonication. La protéine d'intérêt est immuno-précipitée, l'ADN sur lequel elle était liée est récupéré et amplifié par PCR avec des amorces choisies sur les régions promotrices d'intérêt.

195
Avantages : - Cette technique utilisée sur cellules vivantes (plus précis qu'un essai "in vitro" par gel shift par exemple) - Elle permet non seulement de visualiser l'occupation des sites sur les promoteurs à un moment voulu, en réponse à des traitements spécifiques, mais aussi les modifications post-traductionnelles des facteurs de transcription d'intérêt suivant les anticorps utilisés. - L'utilisation d'anticorps anti-histones acétylés permet de visualiser l'état général de la chromatine au niveau de promoteurs spécifiques.

196
Grâce à cette méthode, il est possible de récupérer et de purifier des protéines qui interagissent avec des récepteurs, mais aussi des séquences génomiques adjacentes aux facteurs de transcriptions. Après la purification de ces complexes, les séquences nucléotidiques peuvent être clonées, séquencées et analysées.

198
EXERCICES Interprétation de Southern blot
Digestion de l’ADN génomique de plusieurs individus

199
Interprétation des résultats considérant 2 allèles
Sites Eco R1 4Kb 2Kb Sonde 4.2kb Site polymorphe sur le 2 ième allèle :

200
4.2Kb 4kb 2Kb

201
Interprétation des résultats considérant 2 allèles
Sites Eco R1 4Kb 2Kb Sonde 4.2kb Site polymorphe sur le 2 ième allèle :

202
Interprétation du résultat
Des individus sont 4/2 : 4/ 2 (homozygotes) Des Individus ne montrent aucune hybridation -- Délétion d’au moins 6 kb (homozygote) Le dernier : 4/2 : 4.2/2 : ses deux allèles sont différents ( hétérozygote)
 Des Individus ne montrent aucune hybridation. -- Délétion d’au moins 6 kb (homozygote) Le dernier : 4/2 : 4.2/2 : ses deux allèles sont différents ( hétérozygote)")
203
Diagnostic de la drépanocytose
Maladie génétique fréquente en Afrique noire au Moyen Orient et en Inde . Maladie récessive Mutation ponctuelle par substitution Glu > Val sur le gène beta globine ( codon 6) GAG > GTG Cette mutation supprime un site de restriction pour l’enzyme Mst II
 GAG > GTG. Cette mutation supprime un site de restriction pour l’enzyme Mst II.")
206
LE NORTHERN BLOT // Southern BLOT Isolement de l’ARN colonne oligo dT
Electrophorèse sans dénaturation Hybridation avec une sonde ADN simple brin

207
Interprétation du résultat
Des individus sont 4/2 // 4/ 2 (homozygotes) Des Individus ne montrent aucune hybridation -- Délétion d’au moins 6 kb (homozygote) Le dernier : 4/2 // 4.2/2 : ses deux allèles sont différents ( hétérozygote)
 Des Individus ne montrent aucune hybridation. -- Délétion d’au moins 6 kb (homozygote) Le dernier : 4/2 // 4.2/2 : ses deux allèles sont différents ( hétérozygote)")
208
Diagnostic de la drépanocytose
Maladie génétique fréquente en Afrique noire au Moyen Orient et en Inde . Maladie récessive Mutation ponctuelle par substitution Glu > Val sur le gène beta globine ( codon 6) GAG > GTG Cette mutation supprime un site de restriction pour l’enzyme Mst II
 GAG > GTG. Cette mutation supprime un site de restriction pour l’enzyme Mst II.")
Présentations similaires












>")

 Obtention de l’ADN recombinant>")
 Obtention de l’ADN recombinant>")













