Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Le séquençage à grande échelle au Genoscope
Stratégies actuelles et perspectives P. Wincker, Séminaire INRA, Paris,

2
Status: Public Institute
Mission : provide high-throughput sequencing data to the French Academic community , and carry out in-house genomic projects Creation 1997 Part of the CEA Institut de Génomique since 05/2007

3
Procedures on Scientific Projects
in house : evaluated by the Scientific Committee collaborative: proposed by external labs (annual call for proposals) - evaluated by the Scientific Committee supported by Genoscope's budget shared cost: consumables and labor supported by applicant - other costs on Genoscope's budget approval by Scientific committee > reads paid services
 - evaluated by the Scientific Committee - supported by Genoscope s budget. shared cost: - consumables and labor supported by applicant - other costs on Genoscope s budget - approval by Scientific committee > reads. paid services.")
4
Breakdown of sequencing activity since 1998
Total reads

5
Breakdown of sequencing activity in 2006 Total reads 12 138 976
Coûts partagés 1,4%

6
Successful applications since 1998
Total 188

7
Sequence categories

8
Genomes (finished and in progress)
")
10
L’organisation du séquençage au Genoscope

11
Personnel (01/01/06) 11 7 43 9 5 26 3 6 16 140 2001 Mapping
3 6 16 140 2001 Mapping Libraries, subcloning Sequencing + template prepping Finishing Development Research projects R and D Robotics Informatics Bio-informatics QC and QA Infrastructure (Kitchen, building etc.) TOTAL (FTE) 8 11 18 15 4 27 3 21 24 2 9 150
 TOTAL (FTE)")
12
Niveaux d’accès aux capacités du Genoscope par Appel d’Offres
Projet Séquençage Sanger, 454 (2007), Solexa (2008) Assemblage, finition, clustering Annotation procaryote (MAGE) Annotation eucaryote (GAZE)
, Solexa (2008) Assemblage, finition, clustering. Annotation procaryote. (MAGE) Annotation eucaryote. (GAZE)")
13
Sélection des projets Appel d’offres évalué annuellement par un conseil scientifique externe ( ) A partir de 2008 : Appel d’offres (GIS Ibisa) Projets ANR (Programme Génomique)
 Projets ANR (Programme Génomique)")
14
Sequencing equipment total capacity
ABI (30 M bases/day) 454/GSFLX 1 (100 M bases/day)
 454/GSFLX 1 (100 M bases/day)")
15
Impact des nouvelles technologies de séquençage

16
Evaluation des NTSs au Genoscope
Qualité : des lectures et des assemblages Applications : fonction de la taille des génomes, complémentarité aux autres technologies Impact sur l’obtention d’une séquence «finie »

17
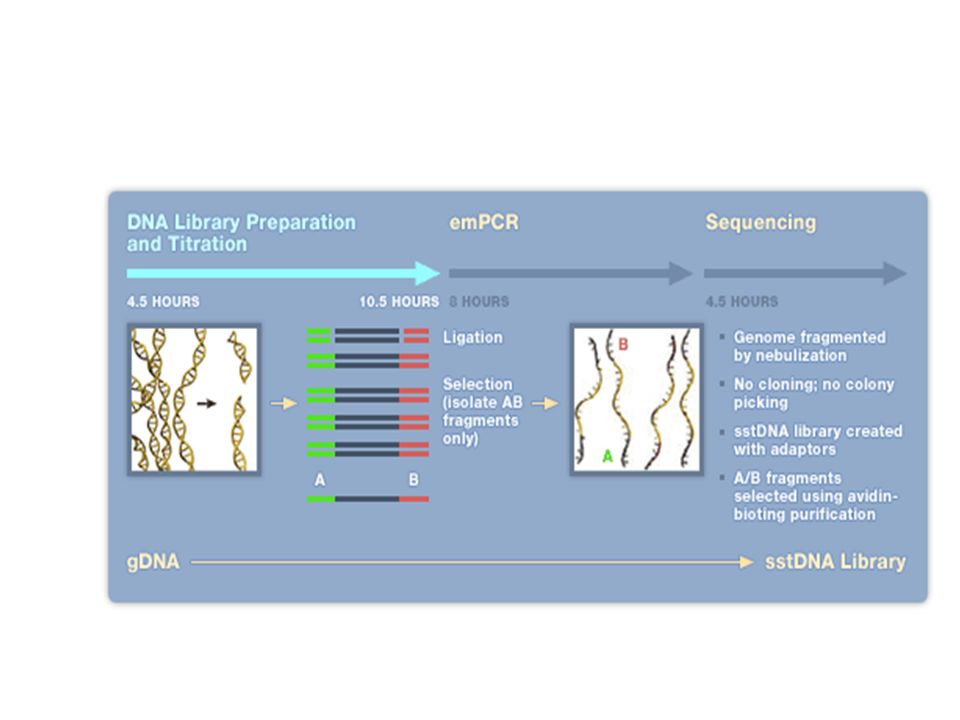
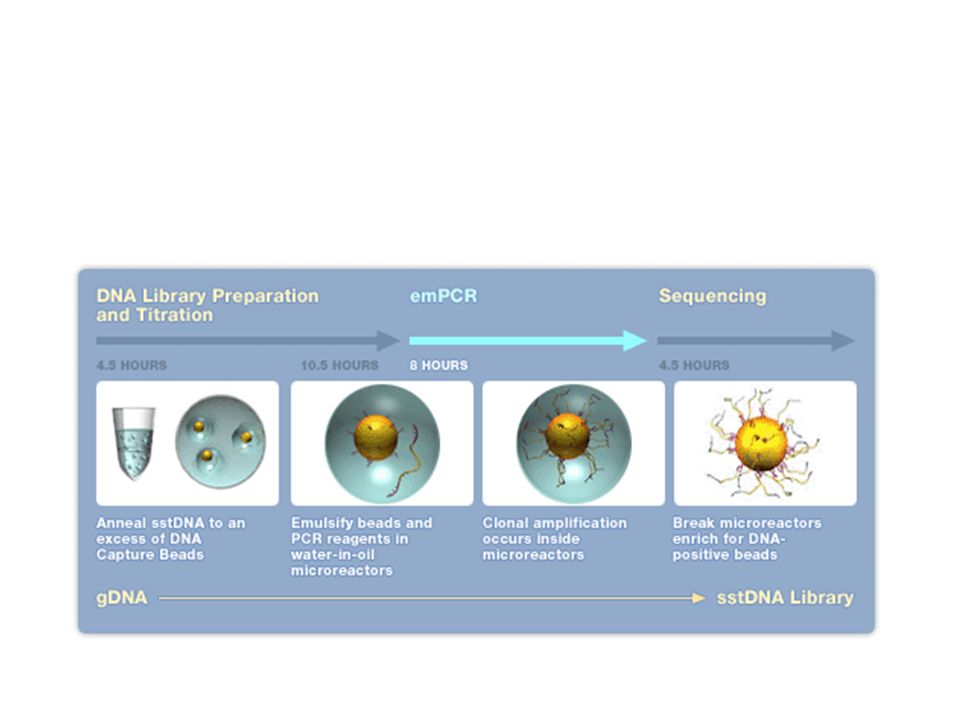
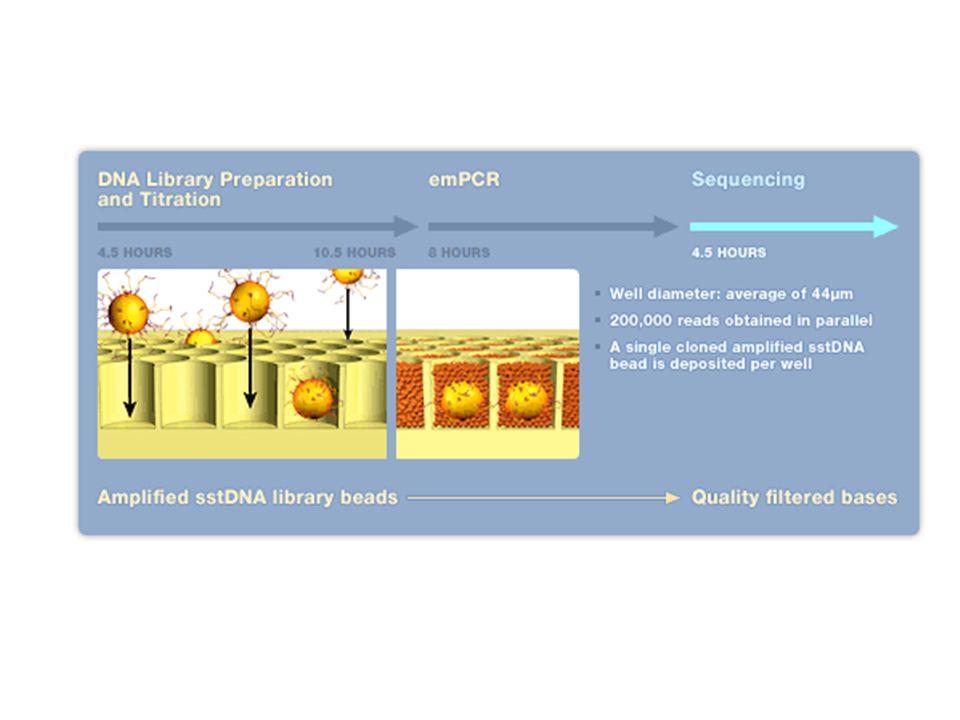
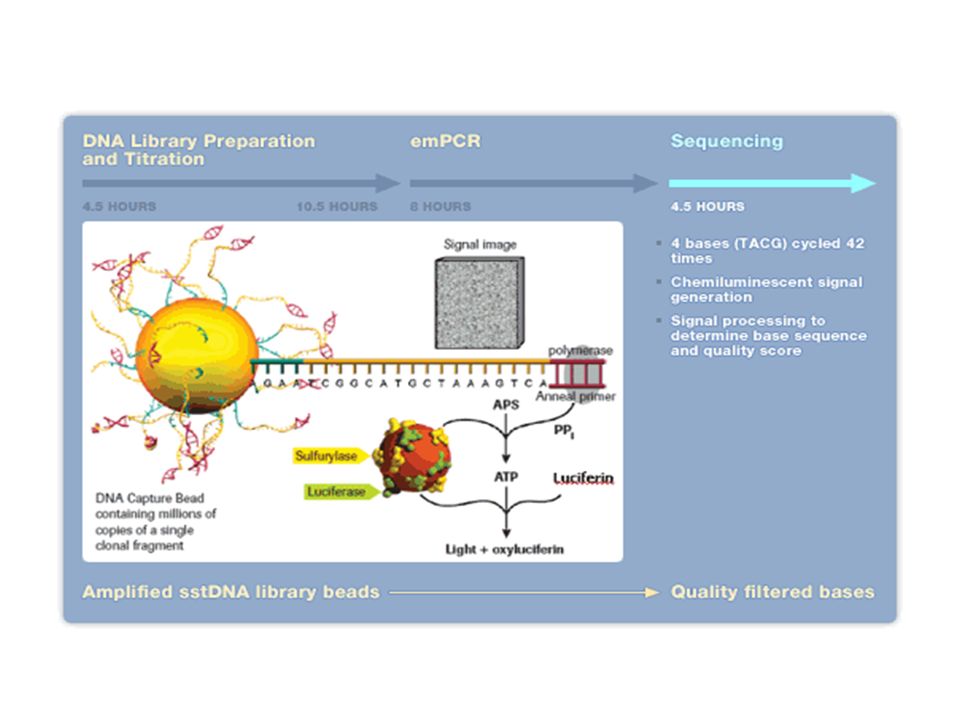
Exemple du séquenceur Roche / 454

23
454 data (flowgram) Sanger data (chromatogram)
 Sanger data (chromatogram)")
24
Evaluation de la qualité des lectures :
Mapping des lectures 454 sur la séquence finie d’Acinetobacter baylyi lectures mappées (soit 99,55%) nt alignés contenant erreurs (soit 1,48% d’erreurs) Avec Q ≥ 20, erreurs (8.10-3) et Q ≥ 40, erreurs (3.10-3) Sur les lectures mappées à 100%, sont sans erreurs (35%)
 nt alignés contenant erreurs (soit 1,48% d’erreurs) Avec Q ≥ 20, erreurs (8.10-3) et Q ≥ 40, erreurs (3.10-3) Sur les lectures mappées à 100%, sont sans erreurs (35%)")
25
Position des erreurs dans les lectures 454

26
Position des erreurs par type dans les lectures 454

27
Evaluation des assemblages 454
Deux types d’assemblage proposés : De novo Dirigé (en utilisant la séquence d’un génome très proche)
")
28
Taille du N50 à différentes profondeurs (assemblage de novo)
")
29
Taille du N50 à différentes profondeurs (de novo vs dirigé)
")
30
Erreurs concentrées dans les régions homopolymériques
Fonction de la taille de l’homopolymère Pour M. agalactiae, couverture de 30x si (N)n avec n<5, taux d’erreur ~1% si (N)n avec n<9, taux d’erreur ~5% Le taux d’erreur dépend de la fréquence des régions homopolymériques Ce n’est pas une valeur absolue
n avec n<5, taux d’erreur ~1% si (N)n avec n<9, taux d’erreur ~5% Le taux d’erreur dépend de la fréquence des régions homopolymériques. Ce n’est pas une valeur absolue.")
31
Evaluation des NTSs au Genoscope
Qualité : des lectures et des assemblages Applications : fonction de la taille des génomes, complémentarité aux autres technologies Impact sur l’obtention d’une séquence «finie »

32
De l’assemblage 454 au génome fini
Points positifs : Pas de clonage présence des régions incompatibles avec E. coli Quasi-insensibilité aux biais compositionnels Vitesse : une semaine de l’ADN à la séquence Points négatifs: Pas de liens entre séquences pas de supercontigage Taux d’erreur élevé dans les homopolymères pas d’assemblage des séquences répétées

33
Microbial Genome Sequencing
Until December 2006 : 12x coverage with Sanger technology, 3 libraries (insert sizes 3 kb, 10 kb, 40 kb) From january 2007 : 4x Sanger coverage, single library (10 or 40 kb) + 20x coverage GS20 reads Assembly with Arachne (Broad Institute) using Sanger reads and Newbler contigs From June 2007, 4x Sanger coverage, single library (10 or 40 kb) , + 15x coverage GSFLX reads Assembly with Arachne (Broad Institute) using Sanger reads and Newbler contigs or with Newbler2 using Sanger reads and GSFLX reads
 From january 2007 : 4x Sanger coverage, single library (10 or 40 kb) + 20x coverage GS20 reads. Assembly with Arachne (Broad Institute) using Sanger reads and Newbler contigs. From June 2007, 4x Sanger coverage, single library (10 or 40 kb) , + 15x coverage GSFLX reads. Assembly with Arachne (Broad Institute) using Sanger reads and Newbler contigs or with Newbler2 using Sanger reads and GSFLX reads.")
34
Le séquenceur Solexa / illumina 1G
Amplification directe sur lames (pas de PCR en émulsion) Séquençage par terminateurs reversibles Longueurs de lecture : bases Débit : lectures / run
 Séquençage par terminateurs reversibles. Longueurs de lecture : bases. Débit : lectures / run.")
35
Applications du Solexa/Illumina 1G (ou ABI Solid)
SNP detection ChIp-Seq Quantitative / qualitative transcriptomics small RNAs …

36
Méthodes pour le re-séquençage :
environnement informatique Objectif : aligner chaque lecture à une localisation unique (si elle existe) sur le génome de référence Exemple si utilisation de blast : 1 lecture contre 140Mb (chr9 humain) ~ 18s/CPU 1 lecture contre 3Gb ~ 386s/CPU 1Gb lectures Solexa contre 3Gb ~ 490 années/CPU 20x de lectures Solexa contre 3 Gb ~ années/CPU Nécessité d’utiliser des méthodes différentes qui tiennent compte de la petite taille des lectures : phageAlign : compare chaque lecture avec les k-mers génomique (en triant les k-mers et en exploitant les parties communes des préfixes pour réduire le travail) ELAND : place les lectures dans une structure de données et les aligne toutes en même temps
 sur le génome de référence. Exemple si utilisation de blast : 1 lecture contre 140Mb (chr9 humain) ~ 18s/CPU. 1 lecture contre 3Gb ~ 386s/CPU. 1Gb lectures Solexa contre 3Gb ~ 490 années/CPU. 20x de lectures Solexa contre 3 Gb ~ années/CPU. Nécessité d’utiliser des méthodes différentes qui tiennent compte de la petite taille des lectures : phageAlign : compare chaque lecture avec les k-mers génomique (en triant les k-mers et en exploitant les parties communes des préfixes pour réduire le travail) ELAND : place les lectures dans une structure de données et les aligne toutes en même temps.")
37
Perspectives d’utilisation Solexa / Illumina 1G
Small RNAs, tags … : avantage de coût par rapport au 454/Roche Séquençage de génomes : attente du développement d’assembleurs adaptés Amélioration de la qualité des séquences 454/Roche assemblés

38
Notions de coût par base (ordre de grandeur)
Sanger (ABI3730xl) : 1000 euros / Mbase taux d’erreur < 99%, assemblage de qualité à ~10 équivalents, supercontigage immédiat Roche/454 GSFLX : 100 euros / Mbase taux d’erreur > 1% dans les régions homopolymériques, assemblage de qualité à ~20 équivalents, pas de supercontigage Illumina 1G : <10 euros / Mbase taux d’erreur <99.9 % , pas d’assemblage de qualité …
 : 1000 euros / Mbase. taux d’erreur < 99%, assemblage de qualité à ~10 équivalents, supercontigage immédiat. Roche/454 GSFLX : 100 euros / Mbase. taux d’erreur > 1% dans les régions homopolymériques, assemblage de qualité à ~20 équivalents, pas de supercontigage. Illumina 1G : <10 euros / Mbase. taux d’erreur <99.9 % , pas d’assemblage de qualité …")
39
4x 15x 0.5x 10-100x Assemblage, finition Assemblage, finition 15x

40
Evolution accélérée des NTSs
Roche / 454 2006 : 20 Mb par run (100 bases par lecture) 2007 : 100 Mb par run (250 bases par lecture) 2008 : 1 Gb par run (500 bases par lecture) Solexa/Illumina 1G 2007 : 1 Gb par run (32 bases par lecture) 2008 : 3 Gb par run (50 bases par lecture, lectures en paires) Difficile de prévoir quelle technologie sera utilisée pour séquencer un génome dans 1-2 ans …
 2007 : 100 Mb par run (250 bases par lecture) 2008 : 1 Gb par run (500 bases par lecture) Solexa/Illumina 1G : 1 Gb par run (32 bases par lecture) 2008 : 3 Gb par run (50 bases par lecture, lectures en paires) Difficile de prévoir quelle technologie sera utilisée. pour séquencer un génome dans 1-2 ans …")
41
Vers un séquençage génomique à très bas coût
Dépendra de la capacité à assembler des séquences courtes et peu chères : Développement de lectures « paired-ends » ? Allongement des longueurs utiles de type Solexa ? Baisse des coûts des lectures 454 ? Amélioration spectaculaire des logiciels d’assemblage ? Arrivée d’une nouvelle technologie ?

42
Une perspective très mobile …
Les programmes de comparaison multi-génomes devraient se généraliser La métagénomique connaîtra un développement exponentiel De nombreux projets jugés jusqu’alors trop coûteux deviennent réalisables … Mais toutes ces perspectives nécessitent des progrès pour être envisageables pour des génomes de grande taille

43
Une perspective très mobile …
Les technologies utilisées peuvent devenir caduques très vite Les besoins informatiques augmentent considérablement Risque d’envahissement par des données massives de faible qualité

44
Director : J. Weissenbach
Sequencing coordination : P. Wincker Production Sequencing: J. Poulain Roche / 454 development : C. Cruaud Informatics: C. Scarpelli, V. Vico, V. Anthouard, J. Leseaux Assembly : J.M. Aury

Présentations similaires






![[number 1-100].](/1/172887/big_thumb.jpg "[number 1-100].>")



 388/2006 établissant un plan pluriannuel pour lexploitation durable du stock de sole du golfe de Gascogne : Évaluation du plan mis.>")








 en 2006 – 2007.>")


