Télécharger la présentation
1
Canalopathies Pathologies cardiaques Dr Florence Raybaud Service de Cardiologie CHU Nice

2
Canalopathies Canalopathies

3
QT long TV catécholergiques QT court Brugada + +

4
Syndrome de Brugada Autosomique dominantAutosomique dominant Homme (8/10)Homme (8/10) Age moyen 40 ansAge moyen 40 ans 5/10 0005/10 000 1er cas 19921er cas 1992 Mutation SCN5A canal Na+Mutation SCN5A canal Na+ Brugada = 4% des MS et 20% des MS sur cœur sainBrugada = 4% des MS et 20% des MS sur cœur sain
Homme (8/10) Age moyen 40 ansAge moyen 40 ans 5/ / er cas 19921er cas 1992 Mutation SCN5A canal Na+Mutation SCN5A canal Na+ Brugada = 4% des MS et 20% des MS sur cœur sainBrugada = 4% des MS et 20% des MS sur cœur sain")
5
Syndrome de Brugada ECG : STECG : ST Risque de MS par FV

6
Syndrome de Brugada Type 1 = ST concave Haut ou triangulaire ≥ 2 mm 2dérivat° V1V2 différentiel = IDM, EP … Type 2 = ST en selle ≥ 2 mm 2dérivat° V1V2 Type 3 = ST < 2mm Variable dans le temps et selon EIC => 3e EIC

7
Syndrome de Brugada Type 2 ou 3 => TEST AJMALINE 1mg/kg 1mn => TEST FLECAINE 2mg/kg 10mn Stop test si type 1 ou arythmie ou QRS +130% Sur 500 Brugada avérés Type 1 = 47% Type 2 ou 3 = 23% Normal = 26%

8
Syndrome de Brugada Clinique:Clinique:Asymptomatique, Syncope, énurésie noctune, réveil brutal nocturne MS MS Contexte : repos, fièvre, surtout hommes SVP : 50% + (1à3 ESV 2 sites >200ms)SVP : 50% + (1à3 ESV 2 sites >200ms) Risque de MS : ATCD de symptome (Syncope, MS) Hommes x5.5 Banque de données Brugada>Priori> EckartRisque de MS : ATCD de symptome (Syncope, MS) Hommes x5.5 Banque de données Brugada>Priori> Eckart Surélévation ST + marquée et + souvent SVP+ (+/-) x8 Type 1 x7.7 Surélévation ST + marquée et + souvent SVP+ (+/-) x8 Type 1 x7.7 pas de sur risque si présence de la mutation
SVP : 50% + (1à3 ESV 2 sites >200ms) Risque de MS : ATCD de symptome (Syncope, MS) Hommes x5.5 Banque de données Brugada>Priori> EckartRisque de MS : ATCD de symptome (Syncope, MS) Hommes x5.5 Banque de données Brugada>Priori> Eckart Surélévation ST + marquée et + souvent SVP+ (+/-) x8 Type 1 x7.7 Surélévation ST + marquée et + souvent SVP+ (+/-) x8 Type 1 x7.7 pas de sur risque si présence de la mutation")
9
Syndrome de Brugada Traitement :Traitement : Test pharmacologique : Test pharmacologique :

10
Syndrome de Brugada Traitement et prise en charge (suite)Traitement et prise en charge (suite) Liste de médicament interdits (bloqueur canal Na+) Dépister les apparenté 1er degré (médico-légal) (Nb : Nantes assistante de recherche 0240165714) (Nb : Nantes assistante de recherche 0240165714) DAI : complications 12% élévation de seuil def 27% élévation seuil stim 27% élévation seuil stim 15% pb détection de R 3.75% chocs inappropiés/an (si a$ = 1.5% de choc /an + Sérécor, quinidine si orage rythmique FV
Traitement et prise en charge (suite) Liste de médicament interdits (bloqueur canal Na+) Dépister les apparenté 1er degré (médico-légal) (Nb : Nantes assistante de recherche ) (Nb : Nantes assistante de recherche ) DAI : complications 12% élévation de seuil def 27% élévation seuil stim 27% élévation seuil stim 15% pb détection de R 3.75% chocs inappropiés/an (si a$ = 1.5% de choc /an + Sérécor, quinidine si orage rythmique FV")
11
Syndrome du QT long congénital Autosomique dominante (90%)Autosomique dominante (90%) 1/5000 naissance1/5000 naissance Définition ECG : QTc > 440 msDéfinition ECG : QTc > 440 ms QT corrigé = QT/√RR (formule de Bazett). Autres anomalie ECG : anomalie de T, torsades de pointe, FV, bradycardie sinusale, BAV2/1 néonatal.Autres anomalie ECG : anomalie de T, torsades de pointe, FV, bradycardie sinusale, BAV2/1 néonatal. 1 2 3 1 fin C. épic 2 fin C. endo. 3 fin C M

12
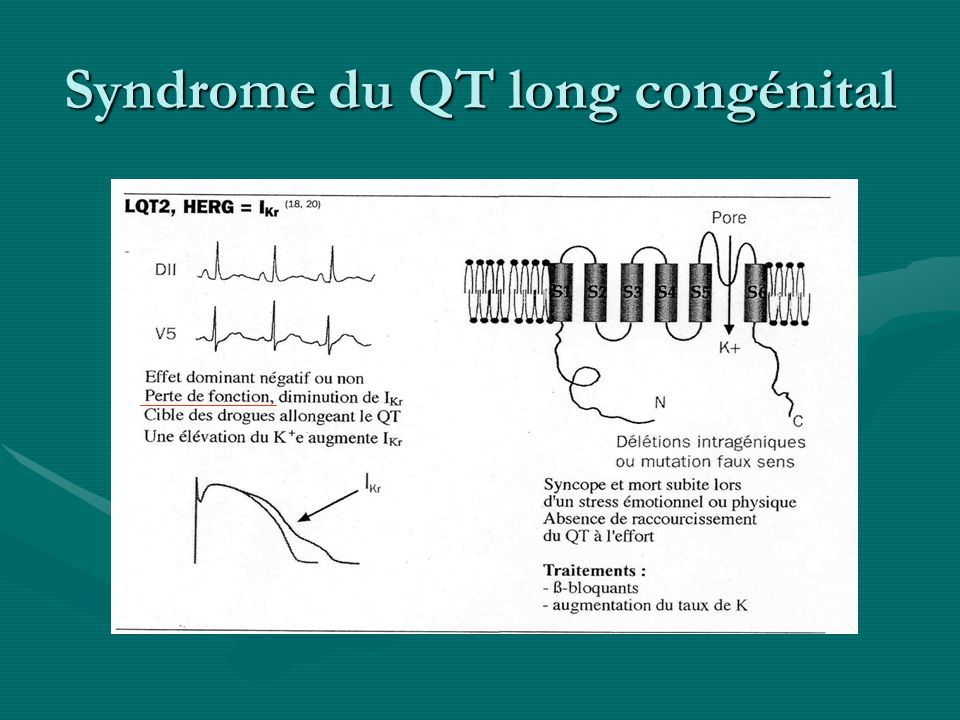
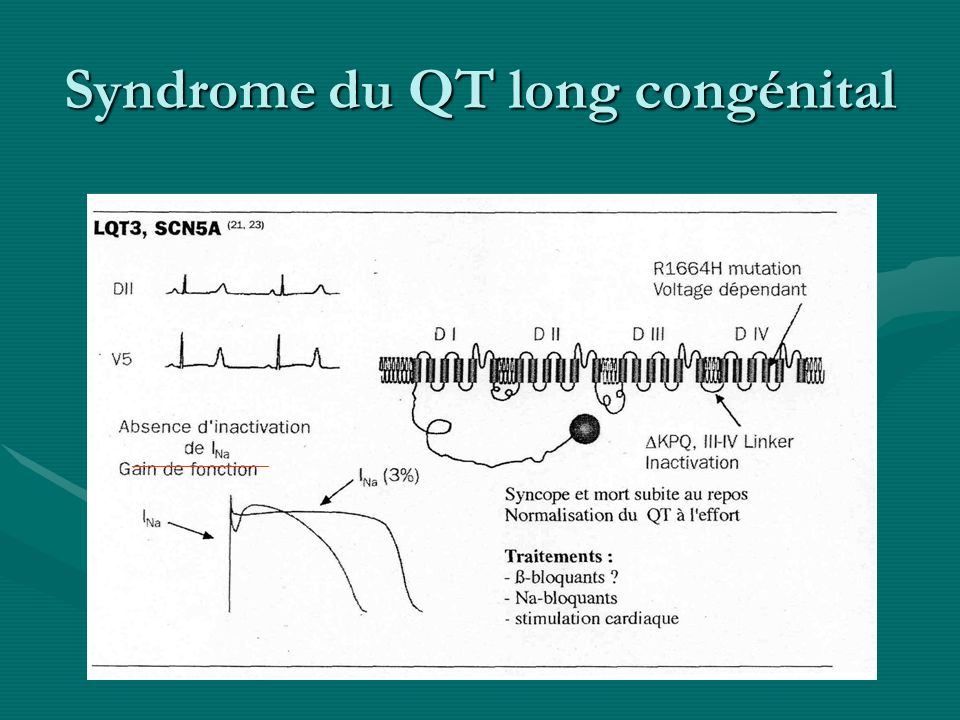
Syndrome du QT long congénital Syndrome Gène Canal%détect°Syndrome Gène Canal%détect° LQT (syndrome de Romano-Ward) LQT1 KCNQ1IKs/KvLQT1 35-45%LQT1 KCNQ1IKs/KvLQT1 35-45% LQT2 KCNH2(Herg)IKr 30-40%LQT2 KCNH2(Herg)IKr 30-40% LQT3 SCN5AINa 5-10%LQT3 SCN5AINa 5-10% LQT4 AnkyrineBNa/Ca <1%LQT4 AnkyrineBNa/Ca <1% LQT5 KCNE1IKs/IsK(minK) <1%LQT5 KCNE1IKs/IsK(minK) <1% LQT6rIKr <1%LQT6rIKr <1% Jerwell et Lange-Nielsen ( + surdité : autosomique récessif) JLN1 KCNQ1IKs 80%JLN1 KCNQ1IKs 80% JLN2 KCNE1IKs/IsK(minK 20%JLN2 KCNE1IKs/IsK(minK 20% Andersen* KCNJ2IK1 50% Timothy ** CACNA1CICaL * paralysie périodique, an morpho ** syndactylie, retard mental
 LQT1 KCNQ1IKs/KvLQT %LQT1 KCNQ1IKs/KvLQT % LQT2 KCNH2(Herg)IKr 30-40%LQT2 KCNH2(Herg)IKr 30-40% LQT3 SCN5AINa 5-10%LQT3 SCN5AINa 5-10% LQT4 AnkyrineBNa/Ca <1%LQT4 AnkyrineBNa/Ca <1% LQT5 KCNE1IKs/IsK(minK) <1%LQT5 KCNE1IKs/IsK(minK) <1% LQT6rIKr <1%LQT6rIKr <1% Jerwell et Lange-Nielsen ( + surdité : autosomique récessif) JLN1 KCNQ1IKs 80%JLN1 KCNQ1IKs 80% JLN2 KCNE1IKs/IsK(minK 20%JLN2 KCNE1IKs/IsK(minK 20% Andersen* KCNJ2IK1 50% Timothy ** CACNA1CICaL * paralysie périodique, an morpho ** syndactylie, retard mental")
13
Syndrome du QT long congénital Clinique :Clinique : Syncope inexpliquée sur cœur nal (1er symptôme) Arrêt cardiaque ou mort subite chez enfant ou jeune Facteur : adrénergie (stress, émotions, effort, noyade) LQT3 : la nuit (et les efforts aussi) médicaments (cf liste)Facteur : adrénergie (stress, émotions, effort, noyade) LQT3 : la nuit (et les efforts aussi) médicaments (cf liste) Contexte : histoire de syncope ou MS familiale histoire de « comitialité » de QT chez un autre / familleContexte : histoire de syncope ou MS familiale histoire de « comitialité » de QT chez un autre / famille Ergométrie et Holter (jour/nuit) le ST ne diminue pas à l’effortErgométrie et Holter (jour/nuit) le ST ne diminue pas à l’effort
 Arrêt cardiaque ou mort subite chez enfant ou jeune Facteur : adrénergie (stress, émotions, effort, noyade) LQT3 : la nuit (et les efforts aussi) médicaments (cf liste)Facteur : adrénergie (stress, émotions, effort, noyade) LQT3 : la nuit (et les efforts aussi) médicaments (cf liste) Contexte : histoire de syncope ou MS familiale histoire de « comitialité » de QT chez un autre / familleContexte : histoire de syncope ou MS familiale histoire de « comitialité » de QT chez un autre / famille Ergométrie et Holter (jour/nuit) le ST ne diminue pas à l’effortErgométrie et Holter (jour/nuit) le ST ne diminue pas à l’effort")
14
Syndrome du QT long congénital LQT1 : large base LQT2 : T aplatie ou double LQT3 : T pointue LQT4 : sinusoïde

15
Syndrome du QT long congénital LQT1

16
Syndrome du QT long congénital Risque = Après un 1ère syncope : 70%Risque = Après un 1ère syncope : 70% Si LQT asymptomatique : 15% MS ou AC Si LQT asymptomatique : 15% MS ou AC + de risque / temps Femme LQT2 Homme LQT3 - de risque/temps Femme LQT1

17
Syndrome du QT long congénital Traitement :Traitement : 1 - BBloquant ++ Nadolol (Corgard*) 50mg/m² /j (adulte1x/j et enfant en 2x/j) Nadolol (Corgard*) 50mg/m² /j (adulte1x/j et enfant en 2x/j) => Eduquer les patients (compliance++) => Eduquer les patients (compliance++) 2- Liste des médicaments interdits 3 – Pas de sport de compétition (loisir ok si BB) 4 – Enquête familiale (50% atteinte)
 50mg/m² /j (adulte1x/j et enfant en 2x/j) Nadolol (Corgard*) 50mg/m² /j (adulte1x/j et enfant en 2x/j) => Eduquer les patients (compliance++) => Eduquer les patients (compliance++) 2- Liste des médicaments interdits 3 – Pas de sport de compétition (loisir ok si BB) 4 – Enquête familiale (50% atteinte)")
18
Liste des médicaments interdits www.qtdrugs.org 1 Drugs that are generally accepted by the QTdrugs.org Advisory Board of the AzCERT to have a risk of causing torsades de pointes. 1 Drugs that are generally accepted by the QTdrugs.org Advisory Board of the AzCERT to have a risk of causing torsades de pointes. 2 Drugs that in some reports have been associated with Torsades de Pointes and/or QT prolongation but at this time lack substantial evidence for causing torsades de pointes. 2 Drugs that in some reports have been associated with Torsades de Pointes and/or QT prolongation but at this time lack substantial evidence for causing torsades de pointes. 3 Drugs to be avoided for use in patients with diagnosed or suspected congenital long QT syndrome. (Drugs on Lists 1, 2 and 4 should also be avoided by patients with QT syndrome.) 3 Drugs to be avoided for use in patients with diagnosed or suspected congenital long QT syndrome. (Drugs on Lists 1, 2 and 4 should also be avoided by patients with QT syndrome.) 4 Drugs that, in some reports, have been weakly associated with torsades de pointes and/or QT prolongation but that are unlikely to be a risk for Torsades de Pointes when used in usual recommended dosages and in patients without other risk factors (e.g., concomitant QT prolonging drugs, bradycardia, electrolyte disturbances, congenital long QT syndrome, concomitant drugs that inhibit metabolism). 4 Drugs that, in some reports, have been weakly associated with torsades de pointes and/or QT prolongation but that are unlikely to be a risk for Torsades de Pointes when used in usual recommended dosages and in patients without other risk factors (e.g., concomitant QT prolonging drugs, bradycardia, electrolyte disturbances, congenital long QT syndrome, concomitant drugs that inhibit metabolism). Anti-arythmiques, diurétiques, laxatifs, psychotropes, anti-infectieux, anti-allergiques …..
 3 Drugs to be avoided for use in patients with diagnosed or suspected congenital long QT syndrome. (Drugs on Lists 1, 2 and 4 should also be avoided by patients with QT syndrome.) 4 Drugs that, in some reports, have been weakly associated with torsades de pointes and/or QT prolongation but that are unlikely to be a risk for Torsades de Pointes when used in usual recommended dosages and in patients without other risk factors (e.g., concomitant QT prolonging drugs, bradycardia, electrolyte disturbances, congenital long QT syndrome, concomitant drugs that inhibit metabolism). 4 Drugs that, in some reports, have been weakly associated with torsades de pointes and/or QT prolongation but that are unlikely to be a risk for Torsades de Pointes when used in usual recommended dosages and in patients without other risk factors (e.g., concomitant QT prolonging drugs, bradycardia, electrolyte disturbances, congenital long QT syndrome, concomitant drugs that inhibit metabolism). Anti-arythmiques, diurétiques, laxatifs, psychotropes, anti-infectieux, anti-allergiques …...")
19
Syndrome du QT long congénital 5 - DAI : TDP/syncope sous BB (IIa,B) LQT1, LQT2 > 500ms (IIb, B) LQT3 homme (peu de syncope mais très léthal) (IIb, B) Suivi : 1x/an => holter (en activité) => ergométrieSuivi : 1x/an => holter (en activité) => ergométrie FC max < 120-150/mn (sinon augmenter BB)FC max < 120-150/mn (sinon augmenter BB) NB : le Mexitil diminue le QT = pas de preuveNB : le Mexitil diminue le QT = pas de preuve
 LQT1, LQT2 > 500ms (IIb, B) LQT3 homme (peu de syncope mais très léthal) (IIb, B) Suivi : 1x/an => holter (en activité) => ergométrieSuivi : 1x/an => holter (en activité) => ergométrie FC max < /mn (sinon augmenter BB)FC max < /mn (sinon augmenter BB) NB : le Mexitil diminue le QT = pas de preuveNB : le Mexitil diminue le QT = pas de preuve")
20
Syndrome du QT long congénital

23
Syndrome du QT court Autosomique dominantAutosomique dominant Homme = FemmeHomme = Femme Surtout jeunes enfants (qqmois) et adultes jeunesSurtout jeunes enfants (qqmois) et adultes jeunes Génétique : détection 35%Génétique : détection 35% Syndrome ShortQTMutation Canal KSyndrome ShortQTMutation Canal K SQT1 KCNH2 IKr SQT2 KCNQ1 IKs SQT3 KCNJ2 IK1 = gain de fonction (diminution Pot. Action)= gain de fonction (diminution Pot. Action) Définition : QT < 320 ms QTc < 340 ms T pointue T pointue
= gain de fonction (diminution Pot. Action) Définition : QT < 320 ms QTc < 340 ms T pointue T pointue.")
24
Syndrome du QT court

25
Clinique :Clinique : Asympt.., syncope, palpitations, MS, FV, faAsympt.., syncope, palpitations, MS, FV, fa MS familiale, fa familiale idiopathiqueMS familiale, fa familiale idiopathique 1er sympt. : 25% MS 24% fa1er sympt. : 25% MS 24% fa ECG : ESV infundib. à couplage Nal ou courtECG : ESV infundib. à couplage Nal ou court QT ne diminue pas trop /effort (ex/ 240 -> 220ms)QT ne diminue pas trop /effort (ex/ 240 -> 220ms) SVP : PREV basse ++ (1ESV => FV) PREOD basse ++ (ESA=> fa)SVP : PREV basse ++ (1ESV => FV) PREOD basse ++ (ESA=> fa)
QT ne diminue pas trop /effort (ex/ 240 -> 220ms) SVP : PREV basse ++ (1ESV => FV) PREOD basse ++ (ESA=> fa)SVP : PREV basse ++ (1ESV => FV) PREOD basse ++ (ESA=> fa).")
26
Syndrome du QT court Traitement :Traitement :DAI Ou Quinidine (seul trt qui normalise QT, augmente PREV et FV non indutible) => IKr : bonne réponse à Quinidine => IKs : réponse moindre Diagnostic différentiel : Digoxine diminue QT (arrêt et voir si QT se normalise)Diagnostic différentiel : Digoxine diminue QT (arrêt et voir si QT se normalise)
 => IKr : bonne réponse à Quinidine => IKs : réponse moindre Diagnostic différentiel : Digoxine diminue QT (arrêt et voir si QT se normalise)Diagnostic différentiel : Digoxine diminue QT (arrêt et voir si QT se normalise)")
27
TV catécholergiques Autosomique dominantAutosomique dominant => mutation RyRr(ryanodine) 50% Autosomique récessiveAutosomique récessive mutation CASQ2 (calsequestrine) Canal Ca++ => surcharge de Ca++ dans la celluleCanal Ca++ => surcharge de Ca++ dans la cellule Enfants (3,5- - 16,5 ans)Enfants (3,5- - 16,5 ans) Homme = femmeHomme = femme Clinique : Syncope à effort/stressClinique : Syncope à effort/stress « comitialité » d’effort Retard diagnostic++ (épilepsie, spasme, vagal..) Histoire familiale de MS (30%)
 50% Autosomique récessiveAutosomique récessive mutation CASQ2 (calsequestrine) Canal Ca++ => surcharge de Ca++ dans la celluleCanal Ca++ => surcharge de Ca++ dans la cellule Enfants (3, ,5 ans)Enfants (3, ,5 ans) Homme = femmeHomme = femme Clinique : Syncope à effort/stressClinique : Syncope à effort/stress « comitialité » d’effort Retard diagnostic++ (épilepsie, spasme, vagal..) Histoire familiale de MS (30%)")
28
TV catécholergiques ECG : FC 60/mn (bradycarde/enfant)ECG : FC 60/mn (bradycarde/enfant) QTC = Nal Effort : ESV (> 120/mn) +/- fa TV bidirectionnelles +++ Traitement : BBloquant = Nadolol 50mg/m²/j ( 2x/j) (pfs ++ 240mg …bien supporté)Traitement : BBloquant = Nadolol 50mg/m²/j ( 2x/j) (pfs ++ 240mg …bien supporté) DAI ?DAI ? Essai de traitement / canal calcique.. En coursEssai de traitement / canal calcique.. En cours Risque : MS : plus risque si syncope survenue tôt < 20 ans : 30-50% FC mortelle si pas de trtRisque : MS : plus risque si syncope survenue tôt < 20 ans : 30-50% FC mortelle si pas de trt RyRr homme +risque / femme ; symptomes survenus plus tôt Suivi : Holter (FC < 130/mn effort; plus d’ESV répétitive/effort, non polymorph.)Suivi : Holter (FC < 130/mn effort; plus d’ESV répétitive/effort, non polymorph.)
Suivi : Holter (FC < 130/mn effort; plus d’ESV répétitive/effort, non polymorph.).")








>")











