Télécharger la présentation
1
Syndromes myéloprolifératifs
Simon LE HELLO Syndromes myéloprolifératifs

2
La Moelle osseuse

3
Hématopoïèse Moelle osseuse Sang périphérique
cellules souches non quantifiables Blastes < 5% Lignée érythroblastique ~30% Lignée granuleuse ~60% Proportion pyramidale inversée Sang périphérique

4
Classification des syndromes myéloprolifératifs
5 entités physiopathologiques (selon polycythemia Vera Study Group) =Processus monoclonal d’un progéniteur pluripotent Leucémie Myéloïde Chronique Splénomégalie myéloïde ou myélofibrose primitive Thrombocythémie essentielle Maladie de Vaquez Leucémie myelo-monocytaire chronique Diagnostic différentiel Syndrome lymphoprolifératif (LLC) Leucémie aigüe (> 30% de blastes)
 =Processus monoclonal d’un progéniteur pluripotent. Leucémie Myéloïde Chronique. Splénomégalie myéloïde ou myélofibrose primitive. Thrombocythémie essentielle. Maladie de Vaquez. Leucémie myelo-monocytaire chronique. Diagnostic différentiel. Syndrome lymphoprolifératif (LLC) Leucémie aigüe (> 30% de blastes)")
5
Leucémie Myéloïde Chronique « de Philadelphie à Nouméa »
La leucémie myéloïde chronique (LMC) est une prolifération myéloïde monoclonale sans blocage de maturation prédominant sur la lignée granuleuse au niveau médullaire et splénique. Fréquence 7 à 20% de toutes les leucémies (22% en NC en 2005) - incidence de 1 à 2 / hab/an (2 en NC en 2005) - tous les ages, surtout l'adulte entre 30 et 50 ans favorisée par l'exposition au benzène et aux rayons ionisants.
 est une prolifération myéloïde monoclonale sans blocage de maturation prédominant sur la lignée granuleuse au niveau médullaire et splénique. Fréquence. 7 à 20% de toutes les leucémies (22% en NC en 2005) - incidence de 1 à 2 / hab/an (2 en NC en 2005) - tous les ages, surtout l adulte entre 30 et 50 ans. favorisée par l exposition au benzène et aux rayons ionisants.")
6
→Affection clonale acquise: marqueur cytogénétique
LMC : Chr Philadelphie →Affection clonale acquise: marqueur cytogénétique = chr Ph1 = néo-gène de fusion bcr-abl qui code pour une tyrosine-kinase activant le pouvoir oncogène d’abl

7
LMC : diagnostic biologique I
a - NFS caractéristique +++ : → Hyperleucocytose > /mm3 à 1 M/mm3 neutrophiles (30 à 40 %) basophilie ++ et éosinophilie myélémie (30 à 50 %) composée de tous les stades de maturation granuleuse métamyélocytes > myélocytes > promyélocytes > rares blastes sans hiatus de maturation +++ →Autres lignées sanguines Anémie modérée Plaquettes normales ou élevées (si abaissées, craindre une acutisation)
 basophilie ++ et éosinophilie myélémie (30 à 50 %) composée de tous les stades de maturation granuleuse métamyélocytes > myélocytes > promyélocytes > rares blastes sans hiatus de maturation +++ →Autres lignées sanguines. Anémie modérée Plaquettes normales ou élevées (si abaissées, craindre une acutisation)")
8
LMC : diagnostic biologique I

9
LMC : diagnostic biologique II
b - Myélogramme : Le myélogramme est identique au frottis sanguin : une hyperplasie granuleuse (80-90 %) maturation normale éosinophilie et basophilie médullaire inutile au diagnostic, n'est réalisé que pour faire une cytogénétique médullaire
 maturation normale éosinophilie et basophilie médullaire. inutile au diagnostic, n est réalisé que pour faire une cytogénétique médullaire.")
10
LMC : diagnostic biologique III
c - Cytogénétique : le chromosome philadelphie : - quasi-totalité des patients (95%) - non spécifique + LAL Ph+ - recherche effectuée sur la moelle - Suivi de la réponse thérapeutique d – Autres examens biologiques BOM (richesse médullaire ++) PAL effondrées t translocation t(9,22) (q34,q11)
 - non spécifique + LAL Ph+ - recherche effectuée sur la moelle. - Suivi de la réponse thérapeutique. d – Autres examens biologiques. BOM (richesse médullaire ++) PAL effondrées. t translocation t(9,22) (q34,q11)")
11
LMC : diagnostic clinique I
Circonstances révélatrices : début insidieux adulte jeune (âge moyen 45 ans) signes peu caractéristiques : asthénie, amaigrissement, grosse rate →NFS de routine
 signes peu caractéristiques : asthénie, amaigrissement, grosse rate. →NFS de routine.")
12
LMC : Evolution / Complications
Evolution en 3 phases : chronique 3 à 5 ans : ↑ leucocytose Accélérée :↑ basophilie aigue 3 à 6 mois entraînant le décès : transformation blastique (>15% blastes dans le sang) Complications: Anémie Hémorragies / Thromboses Infections
 Complications: Anémie. Hémorragies / Thromboses. Infections.")
13
LMC : Traitement I BUT: rémission complète hématologique (NFS normale) ou rémission complète cytogénétique (disparition du clone Ph1) 1 - CHIMIOTHERAPIES CONVENTIONNELLES a - Hydrea* (Hydroxyurée) > Misulban posologie 50 mg/kg/j - effets secondaires cutanés et muqueux Survie globale à 5ans 32% b – Interferon alpha Efficacité certaine en phase chronique Survie prolongée > 10 2 – GREFFES Autogreffe / Allogreffe → Guérison Intrafamilial et < 50 ans (moins de 20%) sinon mort par GVH Probabilité de survie INF Allogreffe → 5ans 50-59% 38-77% → 7ans 32% 58% Risque de décès précoce 4-6% 20-40%
 > Misulban. posologie 50 mg/kg/j - effets secondaires cutanés et muqueux. Survie globale à 5ans 32% b – Interferon alpha. Efficacité certaine en. phase chronique. Survie prolongée > – GREFFES. Autogreffe / Allogreffe → Guérison. Intrafamilial et < 50 ans (moins de 20%) sinon mort par GVH. Probabilité de survie. INF. Allogreffe. → 5ans % 38-77% → 7ans. 32% 58% Risque de décès précoce. 4-6% 20-40%")
14
Espoir mais maladie résiduelle
LMC : Traitement II 3 – NOUVEAUX TRAITEMENTS : Inhibiteurs de Bcr-Abl STI 571 ou Imatinib GLIVEC® inhibiteur spécifique de la tyrosine kinase bcr-abl posologie 400 mg/j en phase chronique Dans tous les cas et immédiatement Effets indésirables rares 95% de RCH et 87% de RCC même sur les formes acutisées Espoir mais maladie résiduelle Durée de la réponse? 10% de résistance Toxicité au long cours?

15
L’ Histoire de Glivec… 1960 : Identification d’une mutation génétique chez les patients atteints de LMC. Dans les années 80 : Un lien est établit entre le chromosome Philadelphie et la Tyrosine kinase. 1990 : Découverte des inhibiteurs de Bcr-Abl. Sélection de la molécule mère parmi les inhibiteurs de la PKC. 1992 : 1er lot de Glivec synthétisé. 1996 : Activité démontrée dans des cellules modifiées Bcr-Abl chez des souris syngénéiques. Juin 1996 : 1er patient atteint de LMC traité. Juin 1999 : Début des essais de phase II. Juin 2000 : Début des essais de phase III. Mai 2001 : Approbation pour la LMC par la FDA. Août 2002 : FDA accorde le statut d’examen prioritaire à Glivec en tant que traitement de 1ère intention de la LMC à un stade précoce.

16
Autres syndromes myéloprolifératifs Diagnostic différentiel
A - Les myélémies réactionnelles : infections sévères nécroses tissulaires et hémolyses régénérations médullaires post-saignement, post aplasie cancers métastatiques traitement : corticoïdes, adrénaline B - Les hyperleucocytoses sans myélémie liée au tabac : modérée : /mm3 C - Les autres syndromes myéloprolifératifs Maladie de Vaquez Thrombocythémie essentielle Splémomégalie myéloïde Leucémie myélo-monocytaire chronique

17
Maladie de Vaquez La Polyglobulie primitive ou maladie de Vaquez est caractérisée par une prolifération non contrôlée de l’ens des lignées myéloïdes mais surtout de la lignée érythroblastique. Son évolution chronique aboutit à une masse sanguine élevée, dominée par le risque de thromboses et l’évolution terminale vers la fibrose médullaire ou la leucémie aigüe. Le pronostic est sombre en 18 mois si non traité mais 12 ans si traité Diagnostic biologique Évoqué sur la NFS : hématie > 6M/μl, hématocrite >55% et hémoglobine entre 18 et 25 g/dl Confirmation par le VGT / hématies marquées au Cr51 Traitement Saignée ou P32 ou chimiothérapie

18
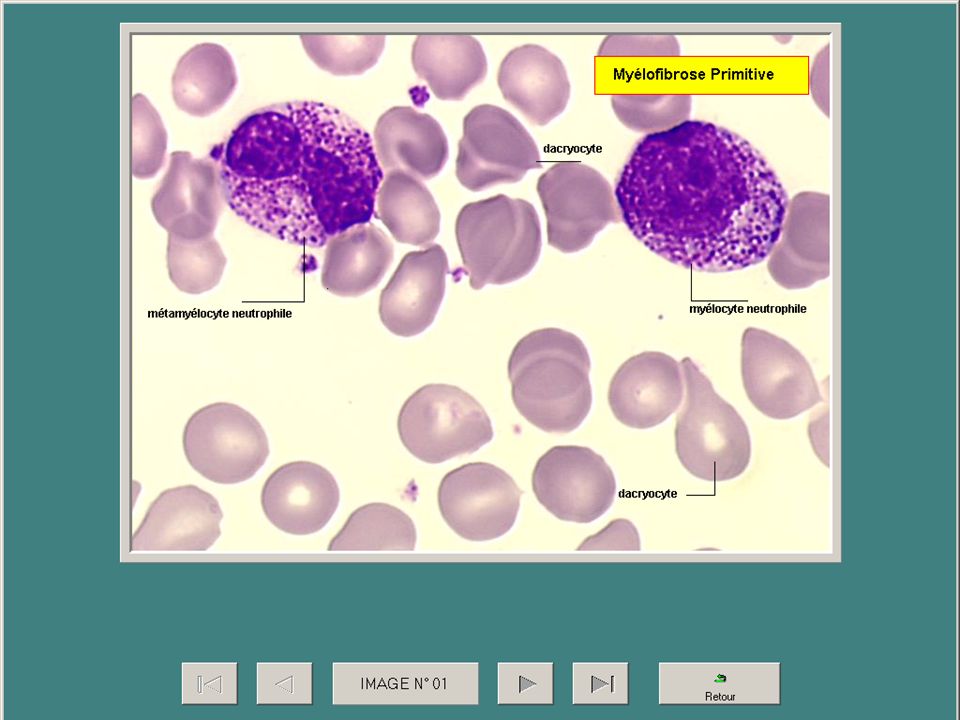
Splénomégalie myéloïde ou myélofibrose primitive
C’est une maladie nettement plus rare que la LMC mais proche avec myélofibrose, survient habituellement après 50 ans. Splénomégalie est le signe majeur avec une rate très volumineuse+++ NFS : érythromyélèmie et dacryocytes+++ BM = myélofibrose et des mégacaryocytes dystrophiques L’évolution est variable de 1 à 15 ans avec transformation en LA (25%) ou pancytopénie Le traitement : Aucun
 ou pancytopénie. Le traitement : Aucun.")
20
Thrombocythémie essentielle (TE)
Excès chronique du nombre des plaquettes >1000 G/L par atteinte monoclonale de la cellule souche mégacaryocytaire, avec une prépondérance chez la femme. NFS : Hyperplaquettose le plus souvent entre 2000 et 5000 G/L. Hyperleucocytose entre 15 et 40 G/L Moelle : hyperplasie mégacaryocytaire dystrophique Complications : Hémorragies/ Thromboses +++ (URGENT) Evolution : survie entre ans max puis transformation en LA Traitement myélosuppressif systématique
 Evolution : survie entre ans max puis transformation en LA. Traitement myélosuppressif systématique.")
21
Leucémie myelomonocytaire Chronique
Double prolifération granulocytaire et monocytaire survenant chez des personnes âgées, d’évolution lentement progressive. Ce syndrome représente 2% des hémopathies. NFS : Monocytose sanguine (>1000G/L de promonocytes) et discrète myélémie. Moelle : hyperplasie myéloïde + infiltration monocytaire (10-15%) Evolution lente voire stable sans traitement chez des sujets âgés
 et discrète. myélémie. Moelle : hyperplasie myéloïde + infiltration monocytaire (10-15%) Evolution lente voire stable sans traitement chez des sujets âgés.")
22
CAS CLINIQUE 1 Homme de 57 ans consulte pour des douleurs épigastiques. A l’auscultation, découverte d’une volumineuse splénomégalie isolée NFP : Hb =13g/dL / plaquettes= 312 G/L / GB= 58 G/L Formule : PNN 70%, myélémie 20%, basophile 6% Myelogramme : moëlle riche avec prédominance de la lignée blanche sans excès de formes immatures. Cytogétique : Chr Ph1 positive LMC

23
CAS CLINIQUE 2 Femme de 69 ans a des douleurs osseuses. Numération avec hyperleucocytose sans splénomégalie et sans signe infectieux. NFP : Hb =13g/dL / plaquettes= 440 G/L / GB= 19,9 G/L Formule : PNN 56%, lympho 20%, mono 23% Myelogramme : moëlle riche sans excès de blastes, lignées neutophiles 64%, erythroblastique 15% et monocyte 15% LMMC

24
CAS CLINIQUE 3





















>")