Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
LEUCEMIE AIGUE CHEZ L’ENFANT.
LEUCEMIE AIGUE LYMPHOBLASTIQUE

2
Définition. Prolifération maligne monoclonale de cellules hématopoïetiques immatures de la lignée lymphoïde

3
Epidémiologie. Incidence 4 pour 100000 par an. Pic 2 à 5 ans.
Sex ratio=1,2 LAL= 5xLAM Registre national des hémopathies chez l’enfant

4
Physiopathologie Remaniements chromosomiques.
Expression aberrante de pro-oncogênes, inhibition des gênes onco suppresseurs Altération de facteurs de transcription. Dysploïdie. Point de départ in utero. Etude de jumeaux monozygote montre que des translocations peuvent apparaître in utero, et que la CSH mutée peut aller d’un jumeau à l’autre dans le placenta. Cependant d’autre accident génétique sont requis pour développer une LA

5
Remaniement chromosomique.

6
Répartition des anomalies différentes entre adultes et enfants
Répartition des anomalies différentes entre adultes et enfants. Mais mécanismes identiques. Plus de LAL B que LAL T 85% de LAL B

7
Hyperdiploïdie > 50 chromosomes associés à un meilleur pronostic
Surexpression de MYC active la transformation cellulaire, et s’associe avec site extra-medullaire dont une atteinte centrale et hyperuricémie.Se voit dans les LAL 3 T(4;11)(q21;q23): LAL secondaire à une chimiothérapie qui utilise inhibiteur de la topoisomérase II. MLL permet de maintenir l’expression de gêne HOX avec activité transcription, réarrangement MLL, gagne des fonction avec une activation de la transcription au dépens de HOX et les cellules s’autorenouvellent. Cliniquement: <2ans, hypercytaire, organomégalie et atteinte du SNC.
(q21;q23): LAL secondaire à une chimiothérapie qui utilise inhibiteur de la topoisomérase II. MLL permet de maintenir l’expression de gêne HOX avec activité transcription, réarrangement MLL, gagne des fonction avec une activation de la transcription au dépens de HOX et les cellules s’autorenouvellent. Cliniquement: <2ans, hypercytaire, organomégalie et atteinte du SNC.")
8
T(12;21)(q13;q22) Tel: facteur de transcription de la famille ETS, essentiel pour le nidation de la CSH ds la Mo; AML facteur transcription de la famille AML1/CBF. Fusion de ces 2 chromosomes responsables d’un transcrit c’est à dire une protéine TEL/AML1 qui va inhiber la transcription qui est normallement initié par AML1. La chromatine qui contient l’ADN est fermé à toute transcription. AML1 forme un hétérodimère avec CBF, se lie à l’ADN( séquence transcriptionnel inductible) via RHD et recruite une protéine avec un activité histone acétyl transférase qui permet d’ouvrir la chromatine puis d’initier la transcription. TEL fusionne avec AML , se lie au séquence core mais au lieux de recruter une activité HAT il s’associe avec une prot déacétylase qui ferme la chromatine et donc inhibe la transcription.
 via RHD et recruite une protéine avec un activité histone acétyl transférase qui permet d’ouvrir la chromatine puis d’initier la transcription. TEL fusionne avec AML , se lie au séquence core mais au lieux de recruter une activité HAT il s’associe avec une prot déacétylase qui ferme la chromatine et donc inhibe la transcription.")
9
Influence des gênes métabolisant les drogues
Déficit homozygote ou hétérozygote en thiopurine transférase: Plus d’effets secondaires Mais meilleur réponse au traitement Absence du génotype de la glutathione S transférase associé à un moindre risque de rechute Thiopurine transférase enz qui catalyse la S-méthylation (inactivation) de la mercaptopurine Pas de phénitoine, phénobarbital ou de carbamazepine, qui augmente l’activité du cytochrome P 450 et donc augmente la clairance des chimio, préférer acide valproïque et gabapentine
 de la mercaptopurine. Pas de phénitoine, phénobarbital ou de carbamazepine, qui augmente l’activité du cytochrome P 450 et donc augmente la clairance des chimio, préférer acide valproïque et gabapentine.")
10
Facteurs étiologiques
Pour les LAL: Trisomie 21 Syndrome de fragilité chromosomique: ataxie télengiectasie Radiations ionisantes. Pour les LAM: Syndrome de fragilité chromosomique (Fanconi, Bloom) Neurofibromatose Anticancéreux (alkylant, inhibiteur des topoisomérase II)
 Neurofibromatose. Anticancéreux (alkylant, inhibiteur des topoisomérase II)")
11
Présentation clinique
Altération de l’état général Syndrome d’insuffisance médullaire: Syndrome anémique Syndrome hémorragique Syndrome infectieux Syndrome tumoral: Adénopathie superficielle voire profonde Splénomégalie. Douleur osseuse lié à l’infiltration de leucémie ds le périoste, idem céphalés Adénopathies fermes, mobiles, non douleureuses

12
Présentation clinique
Complications: CIVD Syndrome de leucostase aigue Localisation neuroméningée (paires craniennes, hypoesthésie de la houppe du menton) Douleurs osseuses. Céphalées Augmentation unilatéral d’un testicule, d’une amygdale. Métaphysaires, nocturnes à la pression des os plats
 Douleurs osseuses. Céphalées. Augmentation unilatéral d’un testicule, d’une amygdale. Métaphysaires, nocturnes à la pression des os plats.")
13
Outils diagnostic. Hémogramme et frottis sanguin (ne pas hésiter à appeler le labo s’il ne vous appelle pas avant!) Myélogramme

14
Diagnostic biologique.
Hémogramme (réticulocyte) et frottis sanguin: Forme typique Leucopénie, ou hyperleucocytose avec une blastose circulante avec hiatus de maturation. Neutropénie. « Hyperlymphocytose » ou lymphopénie. Anémie normochrome normocytaire arégénérative. Thombopénie.
 et frottis sanguin: Forme typique. Leucopénie, ou hyperleucocytose avec une blastose circulante avec hiatus de maturation. Neutropénie. « Hyperlymphocytose » ou lymphopénie. Anémie normochrome normocytaire arégénérative. Thombopénie.")
15
Etapes diagnostiques: faîtes le myélogramme!
1) Etude cytomorphologique. 2) Immunophénotypage. 3) Cytogénétique (biologie moléculaire et caryotype). Immunophénotypage, cytogénétique:
 Etude cytomorphologique. 2) Immunophénotypage. 3) Cytogénétique (biologie moléculaire et caryotype). Immunophénotypage, cytogénétique:")
16
Cytomorphologie Au myélogramme : Classification FAB: Moëlle riche
Diminution des lignées hématopoïetiques normales Blastes>20%: cellules jeunes, chromatine fine, nucléoles importants, cytoplasme +/- abondant Pas de granulations sauf azurophiles, pas de corps d’Auer Classification FAB: LAL1: petit lymphoblaste homogêne LAL2: grand lymphoblaste hétérogène LAL 3: très basophiles (Burkitt like) LAL 1 surtout chez les enfants FAB seulement à but descriptif sans corrélation phénotypique ou cytogénétique.
 LAL 1 surtout chez les enfants. FAB seulement à but descriptif sans corrélation phénotypique ou cytogénétique.")
17
LAL 1: cellule petite dispersée chromatine homogène
LAL 2: gde cellule LAL 3: très basophile.

18
Histologie. Place de la BOM:
Utile en cas d’absence de blastose circulante et moelle inaspirable; Etude de la richesse cellulaire, de l’hématopoïèse résiduelle, de la nécrose. Augmentation de la trame cellulaire et réticulaire. Ne permet pas distinguer une LAL 1 d’une 2. Biopsie de site d’atteinte extra-medullaire( peau, testicule,ganglions) si rechute
 si rechute.")
19
A et b : LAL 1 et 2. C et d : LAL 3, apparence syncitial, cellules plus grosses, chromatine punciforme, nucléole plus petite,, cytoplasme invisible.

20
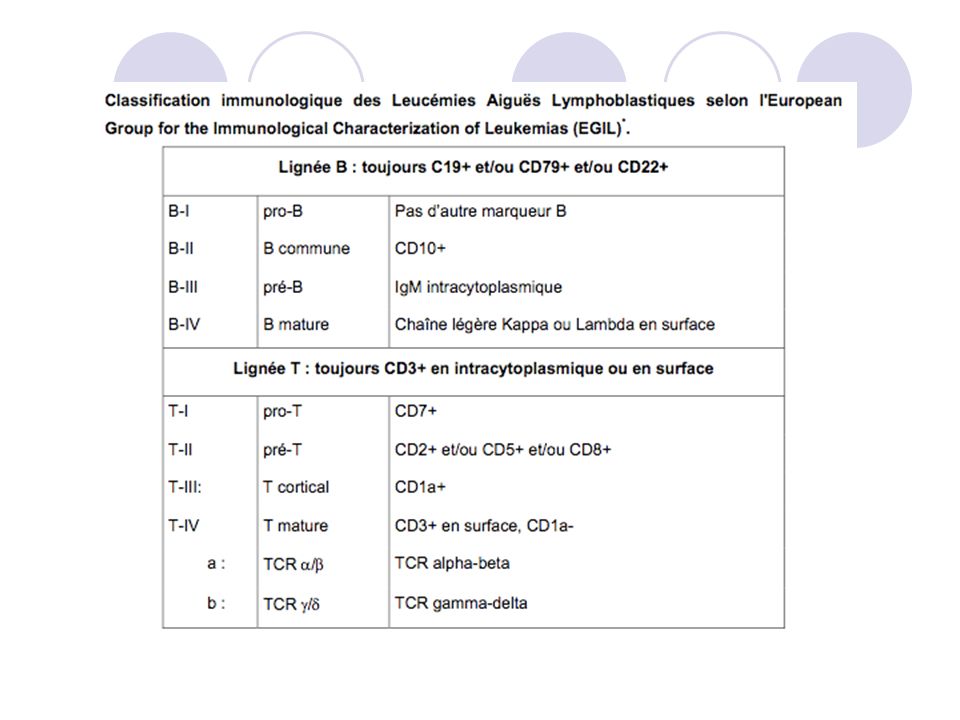
Etape n°2: Immunophénotypage
Etude sur moelle ou sang voire sites extra-médullaires. En immunohistochimie ou par cytométrie en flux. Distingue les lymphoblastes B au T. Ou immunohistochimie. Réaction Sudan black B, MPO

21
Mu est marqueur cytplasmique; sIg: marqueur des Ig de surface.
Classification phénotypique des LAL: 80 % des LAL: LAL B: sous types de LAL semble correspondre à la maturation des lymphocytes B. CALLA- sont de plus mauvais pronostic. Lignée T: les sous types correspondent aux différents stades de maturation des cellules thymiques T La classification OMS ne reconnaît que les stades Précurseur B et T car pas de réelle signification des différents sous-types. Certaines Lymphoblastes peuvent présenter des marqueurs on spécifiques d’une lignée lymphoide; de plus certaines diagnostic différentiel ont le même phénotype que les LAL, ex: thymomes. Donc l’immunophénotypage ne permet pas tous le diagnostic.

23
Etape n°3 : Cytogénétique.
Caryotype et biologie moléculaire Suivi de la maladie résiduelle Avenir: puce à ADN, (DNA microarray) détermine le profil génomique d’une leucémie DNA microarray: d’un échantillon est extrait l’ARN, qui est étudié, pour connaître le profil ADN génomique du tissu. Diagnostic rapide et complet. A permis de démontrer que le LAL qui exprime les marqueurs des lignées T et B représentait une entité particulière. 90% des LAL chez les enfants présentent une anomalies cytogénétiques
 détermine le profil génomique d’une leucémie. DNA microarray: d’un échantillon est extrait l’ARN, qui est étudié, pour connaître le profil ADN génomique du tissu. Diagnostic rapide et complet. A permis de démontrer que le LAL qui exprime les marqueurs des lignées T et B représentait une entité particulière. 90% des LAL chez les enfants présentent une anomalies cytogénétiques.")
25
Hyperdiploïdie > 50 chromosomes associés à un meilleur pronostic
Surexpression de MYC active la transformation cellulaire, et s’associe avec site extra-medullaire dont une atteinte centrale et hyperuricémie.Se voit dans les LAL 3 T(4;11)(q21;q23): LAL secondaire à une chimiothérapie qui utilise inhibiteur de la topoisomérase II. MLL permet de maintenir l’expression de gêne HOX avec activité transcription, réarrangement MLL, gagne des fonction avec une activation de la transcription au dépens de HOX et les cellules s’autorenouvellent. Cliniquement: <2ans, hypercytaire, organomégalie et atteinte du SNC.
(q21;q23): LAL secondaire à une chimiothérapie qui utilise inhibiteur de la topoisomérase II. MLL permet de maintenir l’expression de gêne HOX avec activité transcription, réarrangement MLL, gagne des fonction avec une activation de la transcription au dépens de HOX et les cellules s’autorenouvellent. Cliniquement: <2ans, hypercytaire, organomégalie et atteinte du SNC.")
26
Facteurs de mauvais pronostic cytogénétiques.
BCR/ABL: associé au LAL2

27
Diagnostic différentiel
Affections non malignes: Arthrite chronique juvénile Syndrome mononucléosique PTI Aplasie médullaire Affections malignes: Neuroblastome Lymphome Rhabdomyosarcome

28
Bilan d’extension: Ponction lombaire.
Envahissement extra-médullaire de la leucémie. Facteur de risque: LAL de mauvais pronostic cytogénétique LAL T LAL hypercytaire

29
Définition (children’s cancer group) : plus de 5 GB/l et présence de blastes.
CNS 1 <5GB/l et pas de blastes CNS 2 <5GB/l et présence de blastes CNS 3 ≥5GB/l et présence de blastes, ou atteinte d’un nerf cranien.

30
Bilan des complications
Biologie: CIVD: TP, TCA, fibrinogène, PDF, d-dimères, Fact V Marqueur de lyse tumorale: kaliémie, calcémie, phosphorémie, LDH, acide urique Fonction rénale et hépatique Radio pulmonaire: syndrome de leucostase

31
Bilan préthérapeutique.
Biologie: bilan « standard »: ionogramme, fonction rénale et hépatique Protidémie, albuminémie Groupe sanguin ABO, rhésusx2, sérologie HIV,HBV, HCV, CMV. Typage HLA du patient et de la fratrie. Echographie cardiaque. CECOS

32
Traitement étiologique.
Protocoles+++ Indication dépend du pronostic de la maladie Induction: Préphase de Corticoïdes, Vincristine L-asparaginase Anthracycline doxorubicine ou daunorubicine si LAL de mauvais pronostic (t(9;22);t(4;11)) (LAL 3 traité comme des lymphomes de Burkitt) déxaméthasone+++: meilleure pénétration dans le SNC Vincristine: alcaloïdes de la pervenche, poison du fuseau. L Asparaginase: déplétion en asparagine, inhibition de la synthèse protéique. Athracycline: intercalant: coupures de l’ADN.
;t(4;11)) (LAL 3 traité comme des lymphomes de Burkitt) déxaméthasone+++: meilleure pénétration dans le SNC. Vincristine: alcaloïdes de la pervenche, poison du fuseau. L Asparaginase: déplétion en asparagine, inhibition de la synthèse protéique. Athracycline: intercalant: coupures de l’ADN.")
33
Traitement Prévention neuroméningée:
En prévention des rechutes à localisation neuroméningée A partir de l’induction Chimiothérapie intrathécale: MTX-hemisuccinate d’hydrocortisone-cytarabine. Plus de radiothérapie Localisation neuroméningée que dans 5% des cas. Mais avant l’apparition de la prévention, >80% qui étaient en RC rechutait par méningite leucémique Une étude: évaluation sur 20 enfants à 7 ans de d’une chimio intrathécale: 2 ont un déficit cognitif sur attention et intelligence. Pour la radiothérapie 24 grays associé à des changements à l’IRM MTX: anti folique, AraC (antimétabolite: agit sur bio synthèse de l’ADN)
")
34
2ème phase: consolidation:
MTX haute dose Cytarabine haute dose Etoposide Anthracycline (daunorubicine, doxorubicine) Alkylant (cyclophosphamide, ifosfamide) PL Etoposide: intercalant
 Alkylant (cyclophosphamide, ifosfamide) PL. Etoposide: intercalant.")
35
Place de l’allogreffe Réservée au LAL de mauvais pronostic.
Allogreffe de CSH périphérique ou de cordon? Géno ou phéno identique.

36
3ème phase: traitement d’ entretien.
Injection hebdomadaire de MTX et mercaptopurine per os. Selon protocole: associé à de la vincristine et corticoïdes Durée totale du traitement: 24 mois. Ne pas surdoser en mercaptopurine; ne pas l’administrer dans du lait.

37
Indications et facteurs pronostics
BON pronostic: LAL B Absence de localisation neuroméningée GB<100000/mm3 Hyperdiploïdie Absence de translocation de mauvais pronostic Corticosensibilité Mauvais pronostic: Phénotype indifférencié Localisation neuroméningée T(9;22);t(4;11) Corticorésistance à J8 Absence de RC à l’induction. MRD>10-2 au myelogramme de J35 Selon certains protocoles: <1ans et >10 ans mauvais pronostic
;t(4;11) Corticorésistance à J8. Absence de RC à l’induction. MRD>10-2 au myelogramme de J35. Selon certains protocoles: <1ans et >10 ans mauvais pronostic.")
38
Facteurs de mauvais pronostic cytogénétiques.
BCR/ABL: associé au LAL2

39
Traitement symptomatique.
Transfusion Irradiée. Complication aigue du traitement: Syndrome de lyse cellulaire Infection Thromboembolique Pancréatite Choc anaphylactique. Complication à long terme: cancer secondaire (étoposide), diminution de la fertilité.
, diminution de la fertilité.")
40
Surveillance et évaluation:
La sensibilité de la maladie au traitement doit être évaluée précocément: A J8 après une semaine de corticoïdes: mauvais pronostic si >1000 blastes/mm3 dans le sang A J35-J42: Myélogramme d’évaluation: Rémission complête cytologique et cytogénétique. A J35-42: Evaluation de la maladie résiduelle: grave si >1%

41
Surveillance et évaluation la réponse
Rémission complête est Cytologique: <5% de lymphoblastes dans la moelle avec une récupération de l’hématopoïèse Cytogénétique. Importance du suivi de la maladie résiduelle associée à une meilleure survie sans événement RC cytologique ont toujours des blastes résiduelles

43
Survie globale 80%, EFS à 5 ans: 80%
BCR/ABL: associé au LAL2

44
Quel avenir? Compréhension de la pharmacogénétique

45
Quel avenir?

46
Conclusion Maladie grave. Mais curable! Intérêt des protocoles
Nouveaux outils: puce ADN Avenir passera par la compréhension de toute la cytogénétique.

Présentations similaires



















 Envahissement médullaire puis systématisé par prolifération de cellules hématopoïétiques malignes ETIOLOGIES 1. Idiopathiques Dans.>")

