Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Validation Structurale et Drug Design
Master de Biochimie Validation Structurale et Drug Design Septembre 2006 Copyright UCBL 2006 – Toute reproduction interdite sans le consentement de l’auteur

2
DRUG DESIGN ©

5
Avant tout… Identification d’une cible correspondant à la pathologie
Conception de tests biologiques permettant l’évaluation de l’affinité et de l’activité des molécules synthétisées

6
Technologies Les principales sources de découverte Chimie médicinale
"Ce n'est ni le plus intelligent ni le plus fort qui survivent. C'est le plus adapté." (C. Darwin) Les principales sources de découverte Chimie médicinale Hasard Chimie combinatoire et criblage à haut débit Modélisation moléculaire
 Les principales sources de découverte. Chimie médicinale. Hasard. Chimie combinatoire et criblage à haut débit. Modélisation moléculaire.")
7
La plupart des médicaments connus
La chimie médicinale La plupart des médicaments connus Savoir-faire ancestral Approche basée sur la similitude Points communs à tous les médicaments Concept de "drug-likeness" Règles de Lipinski

8
Approche classique A partir de substances naturelles :
Extraits/broyats Recherche d’activité Synthèse et Production (ex: pénicilline, taxol, cyclosporine…) Constitution d’une bases de données
 Constitution d’une bases de données.")
9
Le hasard Principes actifs naturels Erreurs expérimentales
Paclitaxel, vinorelbine... Cocaïne,morphine,LSD.. Erreurs expérimentales Pénicilline Cisplatine Effets non prévus AZT Viagra Zyban

10
HTS-Chimie combinatoire
Synthèse automatique et rapide de 1000er de molécules Criblage expérimental à haut débit (Hight Throughput Screening) Analyse des informations, tri et évaluation de la diversité moléculaire
 Analyse des informations, tri et évaluation de la diversité moléculaire.")
11
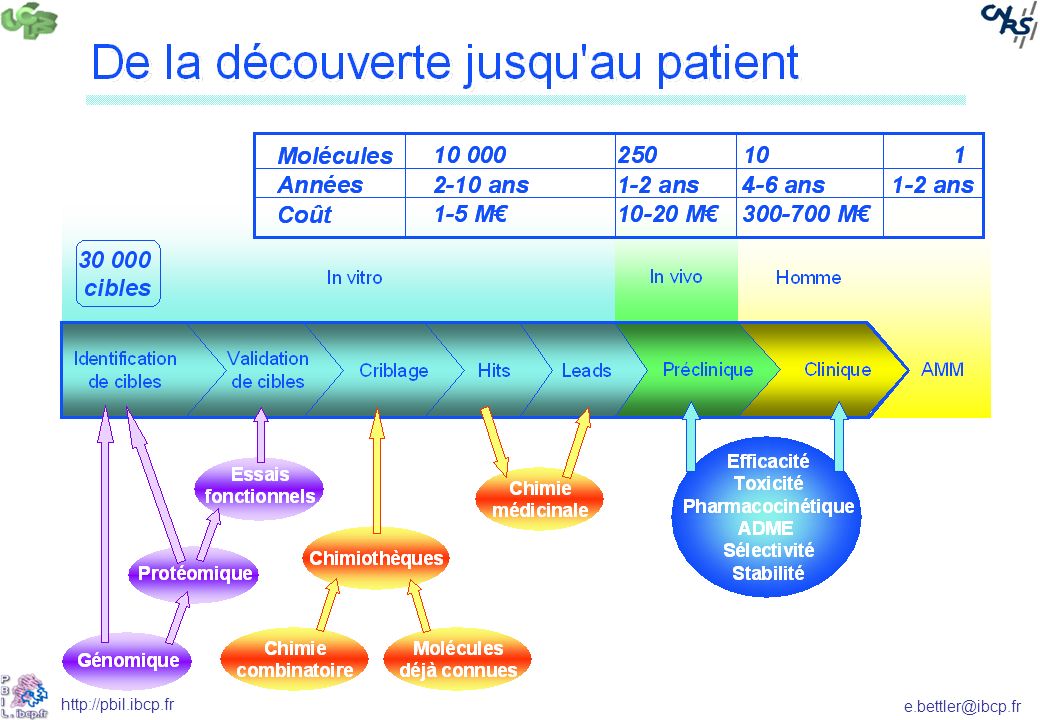
La taille du problème Les médicaments possibles Les médicaments connus

13
Modélisation Moléculaire

14
Cibles et mécanisme d’action de la molécule thérapeutique
Enzymes – inhibiteurs (réversible ou non) Récepteurs – agonistes ou antagonistes Canaux ioniques – bloqueurs Transporteurs – inhibiteurs de transport ADN – agents intercalants, drogue antisens, liants au petit sillon
 Récepteurs – agonistes ou antagonistes. Canaux ioniques – bloqueurs. Transporteurs – inhibiteurs de transport. ADN – agents intercalants, drogue antisens, liants au petit sillon.")
15
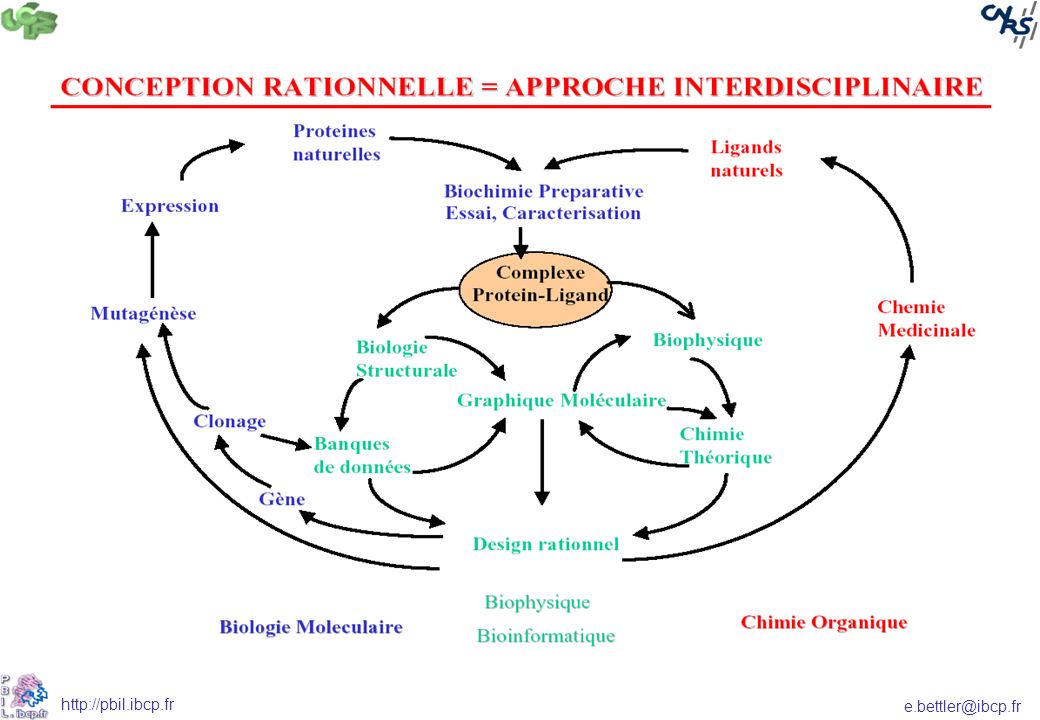
Conception rationnelle
Cible Ligand Connue Inconnue A CHAQUE SITUATION, SES OUTILS…

16
Conception rationnelle
Cible Ligand Connue Inconnue

17
? ? ?

18
Cible et ligand sont connus
« Structure-based drug design » Docking, dynamique moléculaire

19
Docking Largement inspiré de la présentation de G. Schaftenaar
Explain docking is fitting ligand into the receptor, steric and electrostatic match Largement inspiré de la présentation de G. Schaftenaar

20
Objectif Identifier la conformation géométrique correcte du ligand dans son site actif

21
Docking Se divise en 3 étapes : Caractérisation du site actif
Positionnement du ligand dans le site actif Évaluation des interactions entre le ligand et la protéine (définition d’un score)
")
22
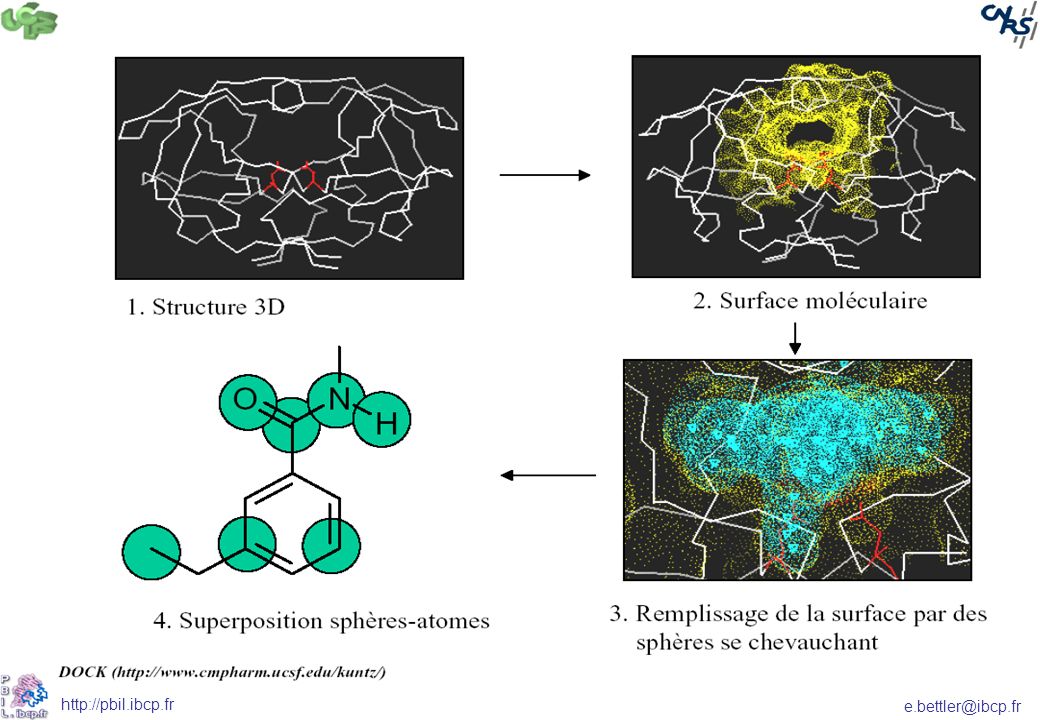
Le site actif Différentes méthodes :
Les cavités de la protéine (récepteur) sont utilisées pour définir une image négative du site actif se composant d’un jeu de sphères se superposant (DOCK) Définition de descripteurs ensuite recherchés à la surface de la protéine descripteurs chimiques (grpt méthyle, aromatique…) ou physico-chimiques (hydrophobicité, potentiel électrostatique)
 sont utilisées pour définir une image négative du site actif se composant d’un jeu de sphères se superposant (DOCK) Définition de descripteurs ensuite recherchés à la surface de la protéine. descripteurs chimiques (grpt méthyle, aromatique…) ou physico-chimiques (hydrophobicité, potentiel électrostatique)")
24
Positionnement du ligand
Le ligand peut être définit comme rigide ou flexible Généralement, le récepteur est toujours définit comme rigide Historiquement, la 1ère approche: tout rigide

25
Ligand rigide La protéine et le ligand sont fixes.
On recherche l’orientation relative des 2 molécules avec la plus basse énergie. On utilise généralement la Transformée de Fourier pour accélérer les calculs (FTDock…)
")
26
Ligand rigide On peut également chercher à évaluer les énergies d’interactions électrostatiques et de VDW pour un complexe Ou encore utiliser des descripteurs (points dans l’espace à qui on assigne certaines propriétés physico-chimique). Les descripteurs du ligand et du récepteur doivent alors coïncidés géométriquement et chimiquement.
. Les descripteurs du ligand et du récepteur doivent alors coïncidés géométriquement et chimiquement.")
27
Complémentarité d’interactions
FlexX

28
Ex. du logiciel FlexX

29
Flexibilité Si on désire introduire la notion de flexibilité du ligand, il faut en plus considérer son espace conformationnel Incorporation des méthodes de: Monte Carlo (FLO98, MCDOCK, AUTODOCK) Dynamique moléculaire (SYBYL) Recuit simulé (AUTODOCK) Algorithme génétique (GOLD, AUTODOCK)
 Dynamique moléculaire (SYBYL) Recuit simulé (AUTODOCK) Algorithme génétique (GOLD, AUTODOCK)")
30
En résumé… Monte carlo (FlexX) « recuit simulé » (AutoDock)
dynamique moléculaire (SYBYL) Algorithmes génétiques (GOLD AutoDock) Géométrie des distances
 Algorithmes génétiques (GOLD AutoDock) Géométrie des distances.")
31
Reconstruction du ligand dans le site actif
Identification d’un ou plusieurs fragments significatifs dans le ligand (ex: cycle d’un aa…) Placement de ces fragments dans le site actif en essayant de maximiser les contacts favorables Chaque orientation de ces fragments est le point de départ d’une étude conformationnelle du ligand entier
 Placement de ces fragments dans le site actif en essayant de maximiser les contacts favorables. Chaque orientation de ces fragments est le point de départ d’une étude conformationnelle du ligand entier.")
32
Flexibilité du ligand

33
Flexibilité du ligand Analyse conformationelle
Insensibilité de la conformation de départ Rapide (1 à 2 min par molécule) Précision insuffisante (rmsd < 2 A dans 75% des cas) Protéine rigide Eau Analyse conformationelle non exhaustive
 Précision insuffisante (rmsd < 2 A dans 75% des cas) Protéine rigide. Eau. Analyse conformationelle non exhaustive.")
34
Scoring

35
Expression de la fonction de scoring
Permet d’estimer la complémentarité ligand-protéine dans les complexes Le plus souvent = estimation du gain d’énergie libre du ligand en interaction avec le récepteur par rapport à la forme libre.

36
Expression de la fonction de scoring
De nombreux faux positifs sont générés mais peuvent être réduits par l’utilisation de fonction de scoring consensus.

37
Exemple de scores Forme et Complémentarité chimique Empirique
Champ de force Base de données Consensus (Cscore)
")
38
Les logiciels DOCK (http://dock.compbio.ucsf.edu/)
FlexX ( GOLD ( AutoDOCK (

39
Conception rationnelle
Cible Ligand Connue Inconnue

40
A partir de la structure du récepteur

41
La cible seule est connue
« Analog-based drug design » Conception de novo, criblage virtuel

42
Conception de novo

43
Conception de novo

44
Conception de novo

45
Assemblage

46
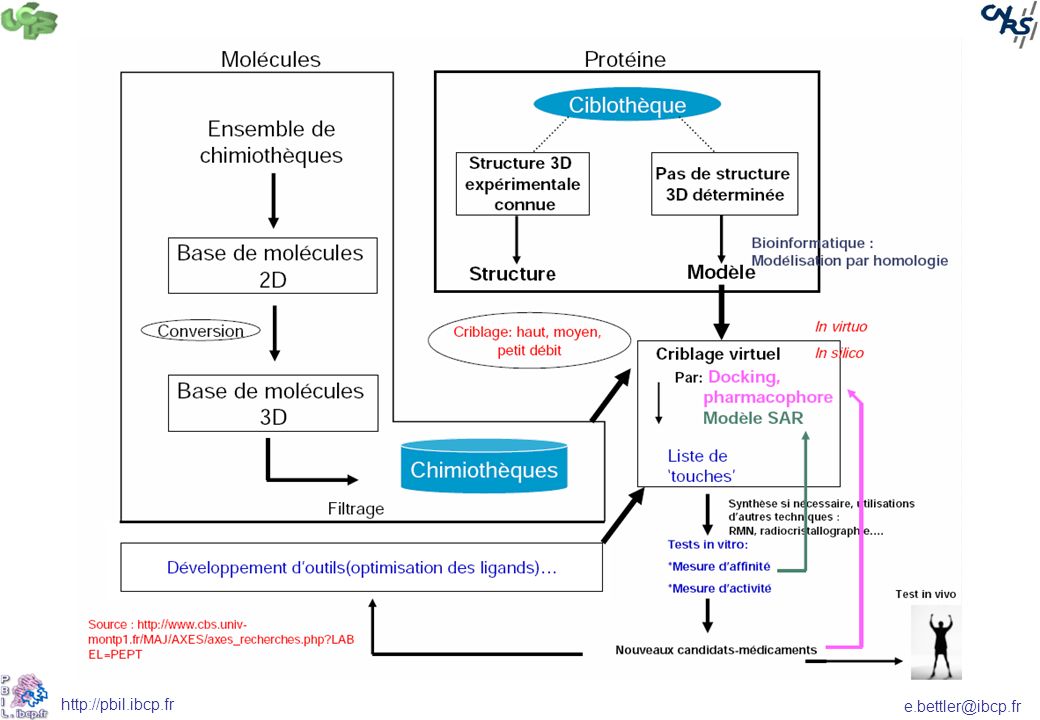
Screening virtuel: <1 sur 100 000
Molécules in silico ~107 ~10 000 molécules Disponibilité, coût, PM, LogP, etc. Enumération des réactions in silico Réactifs ACD Sélection de réactifs 103 à 105 réactifs 108 à 1020 molécules REACTIFS MOLECULES Filtres 1D/2D Tox/réact Règle de 5 Modèles Réaction la plus favorable Docking Sélection finale 2 500 Pharmacophores (3D) Hits Synthèse, HTS
 Hits. Synthèse, HTS.")
47
La règle des 5 de Lipinski
Poids moléculaire < 500 (opt ~= 350) Nbre de liaisons H accepteurs < 10 (opt ~=5) Nbre de liaisons H donneurs < 5 (opt ~=2) -2 < clogP < 5 (opt ~= 3) Nbre d’angles de rotations =< 5 Lipinski et al, Adv. Drug. Del. Rev., 23, 3-25 (1997)
 Nbre de liaisons H accepteurs < 10 (opt ~=5) Nbre de liaisons H donneurs < 5 (opt ~=2) -2 < clogP < 5 (opt ~= 3) Nbre d’angles de rotations =< 5. Lipinski et al, Adv. Drug. Del. Rev., 23, 3-25 (1997)")
48
Conception rationnelle
Cible Ligand Connue Inconnue

49
Le ligand seul est connue
Développement d’un pharmacophore ou d’un modèle QSAR puis criblage virtuel d’une banque de donnée 3D.

50
Pharmacophore Représentation en 3D des propriétés les plus essentielles d’une molécule active

51
Pharmacophores Motif hydrophobe: basé sur les ligands

52
Pharmacophores Motif donneur-accepteur: basé sur le récepteur

53
Criblage sur pharmacophore
Dopamine L = site lipophilique; D = Donneur H; PD = Donneur H protoné

54
Conception rationnelle
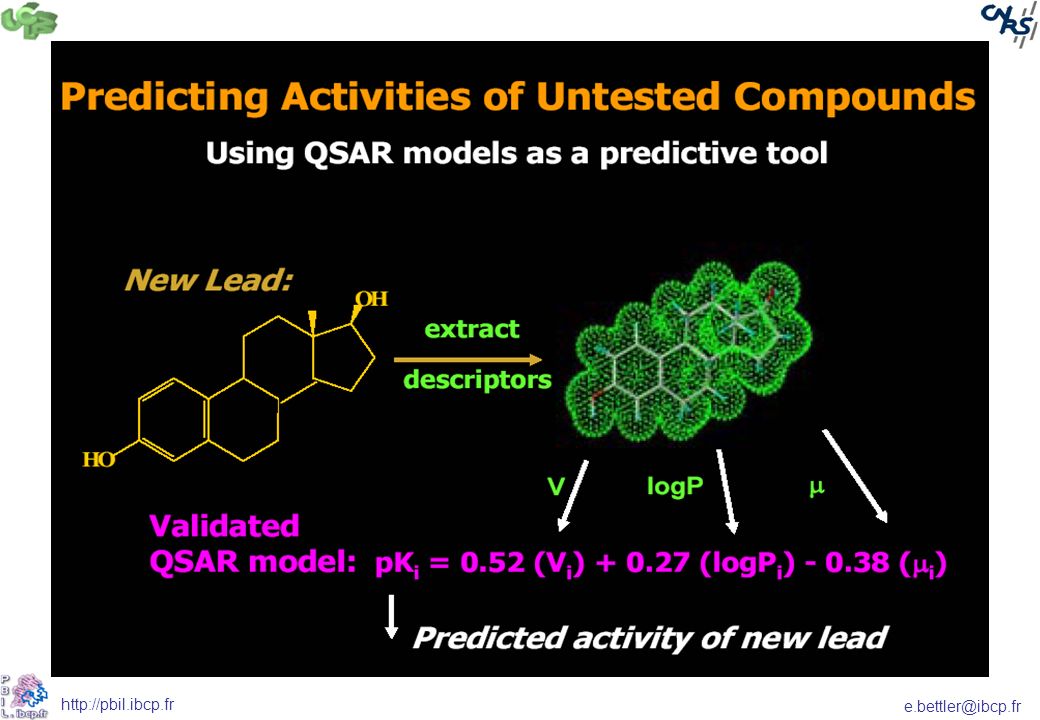
A partir d’un composé naturel, on peut théoriquement synthétiser des millions de dérivés. Nécessité de méthodes quantitatives corrélant paramètres structuraux et activité Quantitative Structure Activity Relationship (QSAR)
")
55
Méthodes de corrélation Quantitative
Association des variations de l'activité biologiques de certaines molécules à leur paramètres structuraux Donne, pour une série chimique donnée et pour une activité définie, une équation de corrélation

56
Equation de corrélation
Permet de déterminer les valeurs des paramètres qui correspondent à une activité maximale et ainsi de prédire l'activité des molécules qui n'ont pas encore été synthétisée La validité d'un modèle QSAR dépendra donc du choix que l'on aura fait sur les paramètres

57
Les paramètres les plus pertinents
Les paramètres associés aux substituants moléculaires Les paramètres associés aux atomes Les paramètres associés à la topologie 1D Les paramètres associés à la topologie 3D

58
Les paramètres associés aux substituants moléculaires
Les paramètres électroniques Les paramètres stériques (encombrement) Les paramètres de lipophilie
 Les paramètres de lipophilie.")
59
Les paramètres électroniques
Une variation de la distribution électronique sur une molécule se traduit par une réactivité chimique différente. De nombreux exemples ont montré qu'une augmentation de la densité électronique conduit à un renforcement de l'activité biologique.

60
Activité insecticide du diéthyl phényl phosphate et du diéthyl 2,4-dicloro-phényl phosphate

61
Les paramètres stériques et lipophiles
Encombrement stérique modifie l’interaction entre une molécule et son récepteur Le caractère lipophile rend compte souvent des propriétés biologiques comme le métabolisme, la distribution dans les tissus, la liaison avec le site récepteur...

62
logP Logarithme du coefficient du rapport 1-octanol/eau (logKOW)
Permet d’estimer la biodisponibilité d’une molécule Équilibre hydrophile/hydrophobe Suffisamment hydrophile pour être soluble dans le sang (eau) Suffisamment hydrophobe pour traverser les membranes cellulaires
 Suffisamment hydrophobe pour traverser les membranes cellulaires.")
63
logP -3 +7 Fortement hydrophile 2 5 Fortement hydrophobe
plupart des molécules thérapeutiques

65
Les paramètres les plus pertinents
Les paramètres associés aux substituants moléculaires Les paramètres associés aux atomes Les paramètres associés à la topologie 1D Les paramètres associés à la topologie 3D

66
Les paramètres associés aux atomes
Le volume atomique Les surfaces atomiques Les charges atomiques partielles L‘électronégativité Les constantes fragmentales de lipophilie

67
Les paramètres les plus pertinents
Les paramètres associés aux substituants moléculaires Les paramètres associés aux atomes Les paramètres associés à la topologie 1D Les paramètres associés à la topologie 3D

68
Les paramètres associés à la topologie 1D
Le volume moléculaire La réfractivité moléculaire Le coefficient de partage La chaleur de formation le potentiel d'ionisation Les constantes d'ionisation

69
Les paramètres les plus pertinents
Les paramètres associés aux substituants moléculaires Les paramètres associés aux atomes Les paramètres associés à la topologie 1D Les paramètres associés à la topologie 3D

70
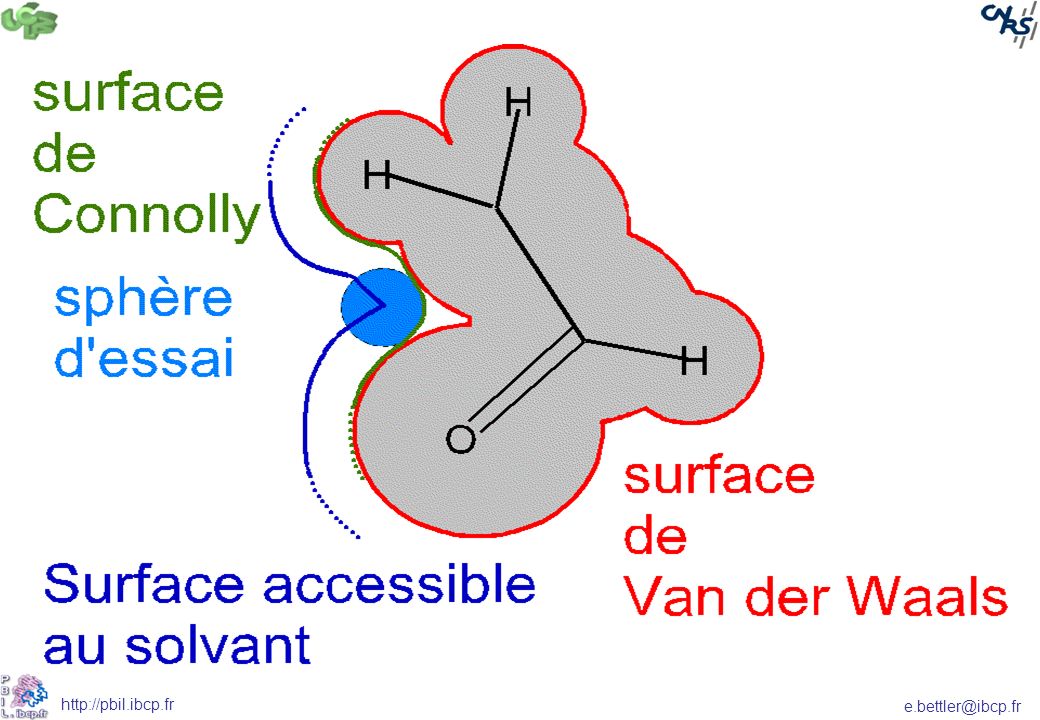
Les paramètres associés à la topologie 3D (3D-QSAR)
La surface moléculaire, accessible au solvant, de connolly ou surface de contact Le potentiel électrostatique (position des groupements chargés) Participation à des liaisons hydrogènes Le potentiel de lipophilie moléculaire Les orbitales moléculaires La forme de la molécule
 Participation à des liaisons hydrogènes. Le potentiel de lipophilie moléculaire. Les orbitales moléculaires. La forme de la molécule.")
72
Les paramètres associés à la topologie 3D (3D-QSAR)
La surface moléculaire, accessible au solvant, de connolly ou surface de contact Le potentiel électrostatique (position des groupements chargés) Participation à des liaisons hydrogènes Le potentiel de lipophilie moléculaire Les orbitales moléculaires La forme de la molécule
 Participation à des liaisons hydrogènes. Le potentiel de lipophilie moléculaire. Les orbitales moléculaires. La forme de la molécule.")
73
Lignes de contour des potentiels électrostatiques

75
Conception rationnelle
Cible Ligand Connue Inconnue

76
La cible comme le ligand sont inconnus
Chimie combinatoire Utilisation d’informations à partir d’autres ligands actifs si connus, analyse de similitude…

77
Conception rationnelle
Cible Ligand Connue Inconnue A CHAQUE SITUATION, SES OUTILS…

79
Quelques réussites falcipain inhibitors
Ring et al. Proc. Natl. Acad. Sci. USA, 90, (1993) FluA HA fusion inhibitors Bodian et al., Biochemistry, 32, (1993) HIV Tat-TAR interaction inhibitors Filikov et al. J. Comput-Aided Mol. Des. 12, (1998) CD4-MHC II inhibitors Gao et al. Proc. Natl. Acad. Sci. USA., 94, (1997) HIV gp41 inhibitors Debnath et al.; J. Med. Chem, 42, (1999)
 FluA HA fusion inhibitors. Bodian et al., Biochemistry, 32, (1993) HIV Tat-TAR interaction inhibitors. Filikov et al. J. Comput-Aided Mol. Des. 12, (1998) CD4-MHC II inhibitors. Gao et al. Proc. Natl. Acad. Sci. USA., 94, (1997) HIV gp41 inhibitors. Debnath et al.; J. Med. Chem, 42, (1999)")
Présentations similaires






Le problème considéré est de démontrer statiquement (à la compilation)>")














 n.>")
