Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Médecine Interne Immunologie Clinique
Mars 2004 Michel Moutschen :

2
Les Déficits Immunitaires

3
Quand faut-il évoquer une immunodéficience?

4
Signes d’appel d’une immunodéficience
8 otites nouvelles en un an 2 sinusites sévères en un an 2 mois d’antibiotiques sans effet 2 pneumonies en un an retard de croissance staturo-pondérale abcès profonds récidivants (peau, organes) muguet persistant ou mycose cutanée âge d’un an recours nécessaire à des antibiotiques IV 2 infections profondes dans la vie (septicémie, méningite, cellulite) histoire familiale de déficit immunitaire
 muguet persistant ou mycose cutanée âge d’un an. recours nécessaire à des antibiotiques IV. 2 infections profondes dans la vie (septicémie, méningite, cellulite) histoire familiale de déficit immunitaire.")
5
Donc, selon les cas, infections
inhabituellement fréquentes inhabituellement longues inhabituellement profondes inhabituellement résistantes aux antibiotiques dans des sites inhabituels avec des germes inhabituels

6
Déficit des barrières Peau et muqueuses
Brèche physique (plaie, cathéter intraveineux) Inflammation locale Chimiothérapie (mucosite) Toxique (tabac) Infection (herpès simplex, influenza,…) Modification des flores locales Antibiotiques (notamment amoxyclav) Corticoïdes en inhalation
 Inflammation locale. Chimiothérapie (mucosite) Toxique (tabac) Infection (herpès simplex, influenza,…) Modification des flores locales. Antibiotiques (notamment amoxyclav) Corticoïdes en inhalation.")
7
Déficits de l’immunité humorale
Surtout infections bactériennes Germes encapsulés Streptococcus pneumoniae Hemophilus influenzae Streptococoques du groupe B Staphylococcus aureus Bacilles entériques gram-

8
Déficits de l’immunité humorale
Infections virales Souvent primo-infections normales en fréquence et en sévérité Déficit de la mémoire à long terme Rougeoles et varicelles à répétition Infections sévères à entérovirus (atteintes neurologiques) Infections parasitaires : giardiases
 Infections parasitaires : giardiases.")
9
Causes des déficits de l’immunité humorale
Phénomènes lymphoprolifératifs Myélome Leucémie lymphoïde chronique B Perte de protéines Syndrome néphrotique Brûlures Entéropathie avec perte de protéines Malnutrition

10
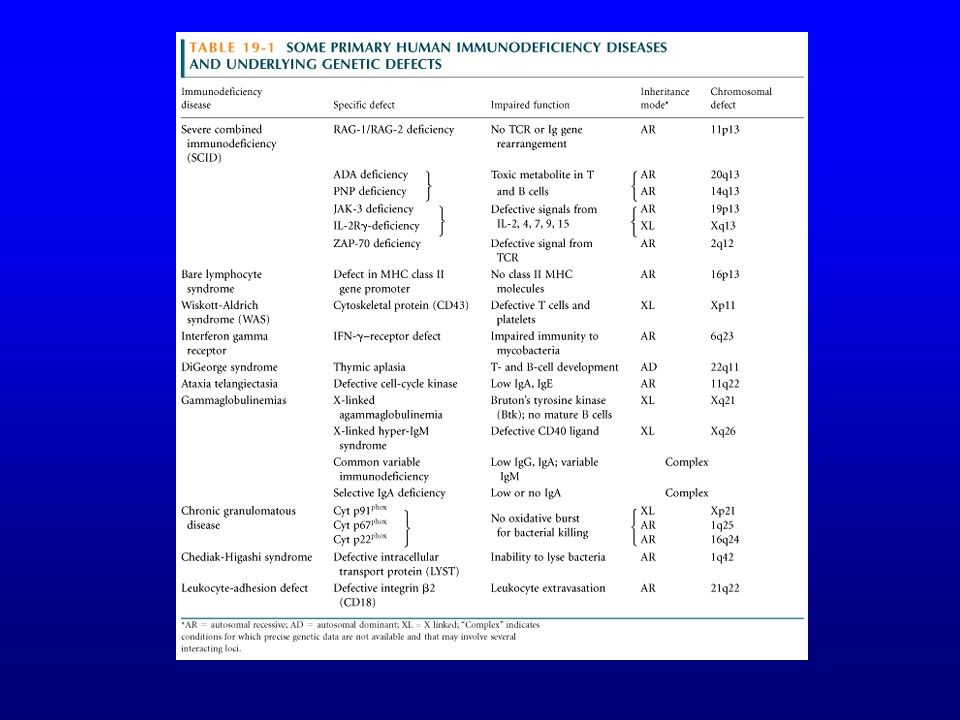
Déficits de l’immunité humorale
Déficits génétiques Maladies autosomales ou liées au chromosome X graves et rares, généralement prises en charge par les pédiatres Maladie de Bruton, agammaglobulinémie hyper IgM, … Hypogammaglobulinémies d’étiologie inconnue avec prédisposition génétique

12
Hypogammaglobulinémies d’étiologie inconnue avec prédisposition génétique
Déficit en IgA Hypogammaglobulinémie commune variable ou immunodéficience commune variable Déficit en sous-classes

13
Hypogammaglobulinémies d’étiologie inconnue avec prédisposition génétique
Moins sévères que les hypogammaglobulinémies monogéniques Plus fréquentes Diagnostic occasionnel tard dans la vie Association à des phénomènes dysimmunitaires

14
Hypogammaglobulinémie commune variable (CVH ou CVI ou CVID)
le plus fréquent des déficits immunitaires primitifs après le déficit isolé en IgA syndrome probablement hétérogène caractérisé par une hypogamma-globulinémie malgré un nombre normal (ou presque) de lymphocytes B hypogammaglobulinémie variable selon les individus et les antigènes considérés (vs Bruton) Fréquemment associé à l’haplotype A1B8DR3
 de lymphocytes B. hypogammaglobulinémie variable selon les individus et les antigènes considérés (vs Bruton) Fréquemment associé à l’haplotype A1B8DR3.")
16
CVI Présentation Tout âge (svt jeunes enfants ou jeunes adultes)
Infections bactériennes récurrentes Germes encapsulés, moraxella Giardiases Hyperactivation du système immunitaire Splénomégalie, syndromes hémophagocytiques, hyperplasie nodulaire de l’intestin, granulomes pulmonaires ou hépatiques Autoimmunité (thyroïde, diabète, vitiligo, pelade)
")
17
CVI Complications tardives
Infections : bronchectasies, cholangites à Campylobacter, arthrites à Ureaplasma urealyticum Malabsorption (entéropathie de type maladie coeliaque) Splénomégalie, hypersplénisme Lymphomes B Autoimmunité Thymome (myasthénie grave, anémie aplastique, PTI)
 Splénomégalie, hypersplénisme. Lymphomes B. Autoimmunité. Thymome (myasthénie grave, anémie aplastique, PTI)")
18
Déficit sélectif en IgA
Fréquent : 1/400 à 1/800, surtout chez les sujets atopiques Déficit au niveau du sérum et des sécrétions Etiologie hétérogène Contexte HLA-A1B8DR3 Une variante du CVI? Facteurs exogènes : chimiothérapie, phénytoïne

19
Déficit en IgA Présentation
Décours souvent bénin (sauf si déficit associé IgG2) Infections ORL fréquentes, plus rarement bronchectasies, diarrhée chronique (giardiases) Phénomènes allergiques (IgE accrues) et autoimmunitaires (LED, PR, JCA, Biermer, maladie coeliaque) Réactions transfusionnelles
 Infections ORL fréquentes, plus rarement bronchectasies, diarrhée chronique (giardiases) Phénomènes allergiques (IgE accrues) et autoimmunitaires (LED, PR, JCA, Biermer, maladie coeliaque) Réactions transfusionnelles.")
20
Déficit en IgA Diagnostic IgA sériques indétectables (<0.05g/l)
Pas d’IgA salivaire, présence d’IgG et d’IgM IgG totales et IgM normales, IgG2 et IgG4 svt abaissées IgE svt accrues Autoanticorps fréquents dont anti-IgA Fonction T normale

21
Déficit sélectif en sous-classes d’IgG
Tout âge IgG2 et/ou IgG4 : la plupart des anticorps anti-polysaccharidiques sont des IgG2 donc déficit associé à infections récurrentes (pneumocoque, Haemophilus influenzae, Pseudomonas aeruginosa), bronchectasies; svt associé à déficit en IgA Importance clinique encore controversée
, bronchectasies; svt associé à déficit en IgA. Importance clinique encore controversée.")
22
Traitement des hypogammaglobulinémies
Immunoglobulines par voie intraveineuse (0.4 à 0.5 g/kg) Toutes les 3 à 4 semaines Viser une concentration résiduelle d’IgG supérieure à 5g/l
 Toutes les 3 à 4 semaines. Viser une concentration résiduelle d’IgG supérieure à 5g/l.")
23
Traitements des hypogammaglobulinémies
Pour les patients avec des valeurs modérément abaissées (entre 3 et 5 g/l IgG) ou pour les déficits en sous-classes Évaluer la fréquence et la gravité des infection Evaluer le titre des anticorps dirigés contre un pathogène donnée (p.ex. anticorps antipolysaccharidiques après vaccin) Les Ig intraveineuses ne sont pas un traitement anodin Risque (faible) d’affection transmissible par le sang Effets secondaires immédiats Coût Hôpital de jour tous les mois
 ou pour les déficits en sous-classes. Évaluer la fréquence et la gravité des infection. Evaluer le titre des anticorps dirigés contre un pathogène donnée (p.ex. anticorps antipolysaccharidiques après vaccin) Les Ig intraveineuses ne sont pas un traitement anodin. Risque (faible) d’affection transmissible par le sang. Effets secondaires immédiats. Coût. Hôpital de jour tous les mois.")
24
Traitement des hypogammaglobulinémies
Les préparations d’immunoglobulines IV ne contiennent que des IgG Elles sont inutiles et souvent dangereuses chez les patients qui ont des déficits en IgA Contre-indication dans les déficits sélectifs en IgA avec présence d’anti-IgA Si nécessaire : préparations pauvres en IgA (donneurs déficients ou traitement in vitro)
")
25
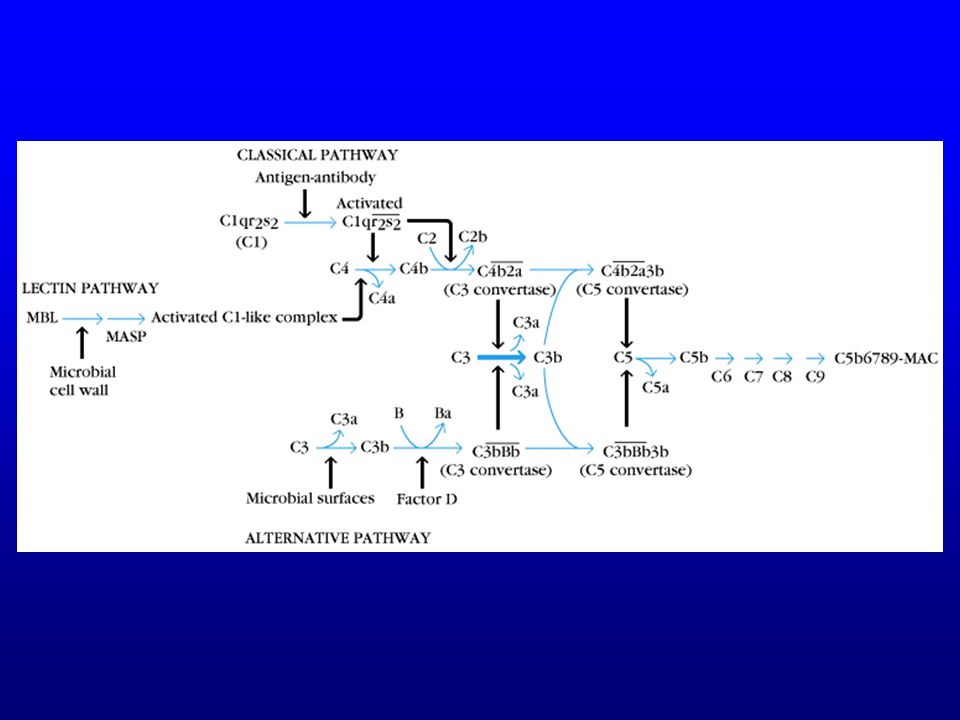
Déficits du complément

26
Conséquences de l’activation du complément

27
Donc deux activités antimicrobiennes du complément :
founir des opsonines (C3b et iC3b) lyser directement les bactéries (Gram-)
 lyser directement les bactéries (Gram-)")
28
Conséquences de l’activation du complément
bactéries Gram-

31
Donc déficit en C1, C2, C4 : peu d’infections
C3 : infections à germes encapsulés (défaut d’opsonisation) C5 à C9 : infections à Neisseria Neisseria gonorrhoeae Neisseria meningitidis
 C5 à C9 : infections à Neisseria. Neisseria gonorrhoeae. Neisseria meningitidis.")
33
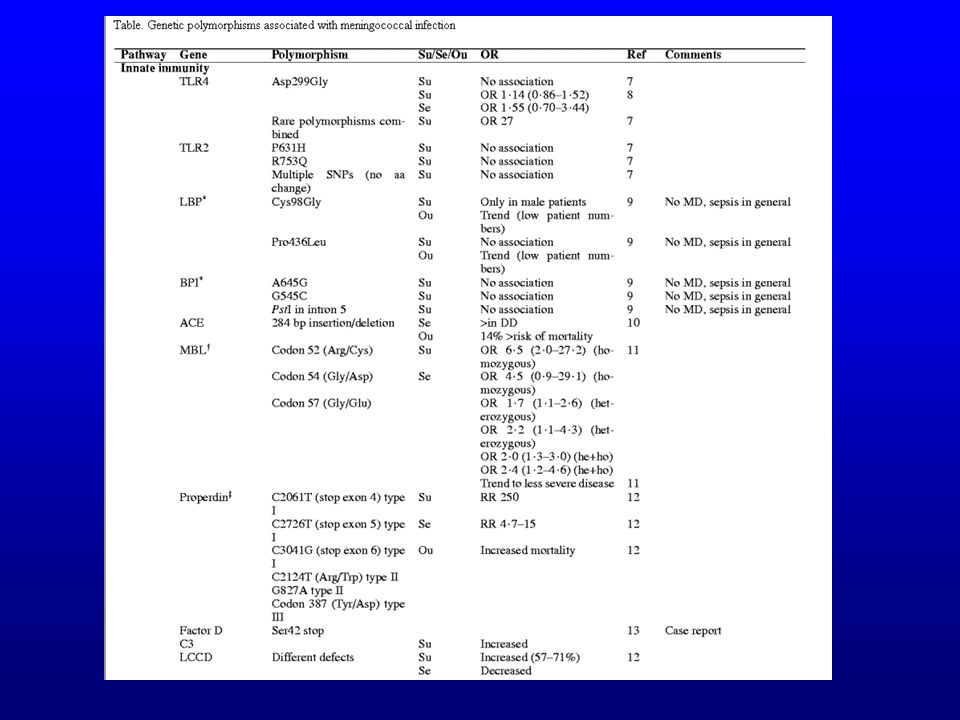
Susceptibilité et sévérité des infections à méningocoques
Déficiences et certains polymorphismes de la properdine et de la MBP

34
Voie alterne Facteur B : une fois fixé sur C3b, devient le substrat du facteur D (protéase équivalent de C1s) C3Bb : C3 convertase de la voie alterne Properdine : augmente la ½ vie de la C3 convertase de la voie alterne (530 minutes)
")
35
Déficit des phagocytes
Germes : E. Coli, Klebsiella pneumoniae, Pseudomonas aeruginosa, enterobacter, serratia, Staph. Epidermidis, Candida, Aspergillus Déficits quantitatifs Neutropénies et agranulocytoses Importance fondamentale (voir cours d’hématologie) Déficits qualitatifs
 Déficits qualitatifs.")
36
Déficits qualitatifs des phagocytes
Acquis Leucoses : LMA, LLA, LMC, LLC, syndromes myélodysplasiques Médicaments : corticoïdes, antimétabolites Atopie : cytokines de type Th2 et fonctions microbicides Syndrome de Job (hyperIgE) Diabète Carence en zinc?
 Diabète. Carence en zinc")
37
Déficits qualitatifs des phagocytes
Génétiques Maladies granulomateuses chroniques Déficit en myélopéroxydase Déficit en récepteur pour iC3b

38
Traitement des déficits phagocytaires
Facteurs de croissance (GM-CSF ou G-CSF) Interféron-gamma Transfusion de granulocytes Prophylaxies
 Interféron-gamma. Transfusion de granulocytes. Prophylaxies.")
39
Asplénisme et splénectomie
Rôle de la rate dans l’élimination des germes du sang mais aussi dans l’initiation des réponses humorales et dans la production de properdine Risque fortement accru S. pneumoniae H. influenzae Neisseria

40
Immunité à médiation cellulaire
Germes intracellulaires parce que seul le système d’échantillonnage des protéines de l’intérieur de la cellule (présentation par des molécules de classe I ou II) permet au système immunitaire de percevoir ces agents qui se cachent dans les cellules (les anticorps ne peuvent les atteindre) les systèmes de microbicidie oxydative des macrophages ont absolument besoin des cytokines de type Th1 (IFN-g, IL-12) pour être activés
 permet au système immunitaire de percevoir ces agents qui se cachent dans les cellules (les anticorps ne peuvent les atteindre) les systèmes de microbicidie oxydative des macrophages ont absolument besoin des cytokines de type Th1 (IFN-g, IL-12) pour être activés.")
41
Immunité à médiation cellulaire
infections mycotiques (candidose mucocutanée,…) infections virales (svt virus latents de la famille herpès) pneumocystoses bactéries intracellulaires (mycobactéries) svt déficits qualitatifs de l’immunité humorale taux d’anticorps normal mais anticorps de mauvaise qualité (rôle des lymphocytes T dans l’immunopoïèse B) Infections qui peuvent toucher le nouveau-né (pas de transfert des lymphocytes T maternels)
 infections virales (svt virus latents de la famille herpès) pneumocystoses. bactéries intracellulaires (mycobactéries) svt déficits qualitatifs de l’immunité humorale. taux d’anticorps normal mais anticorps de mauvaise qualité (rôle des lymphocytes T dans l’immunopoïèse B) Infections qui peuvent toucher le nouveau-né (pas de transfert des lymphocytes T maternels)")
42
Causes des déficits de l’immunité cellulaire
Leucoses et lymphomes : Hodgkin Infections virales (HIV, CMV, EBV, influenza, brucellose,…) Sarcoïdose Carence en zinc Diabète, stress, malnutrition, insuffisance rénale Médicaments : corticoïdes
 Sarcoïdose. Carence en zinc. Diabète, stress, malnutrition, insuffisance rénale. Médicaments : corticoïdes.")
43
Déficits primitifs de l’immunité à médiation cellulaire
Di George = aplasie congénitale thymique délétion d’une partie du chromosome 22 pendant la vie embryonnaire aplasie thymique anomalies cardiaques hypoparathyroïdie dysplasie des oreilles et de la bouche

45
Exploration des déficits immunitaires
screening sang complet et formule hémoleucocytaire dosage immunoglobulines IgG, A, M, D, E Généralement IgG > 5g/l et IgA > 0.5g/l Si valeurs limites, nécessité d’évaluer des réponses spécifiques (p.ex. anti-tétanos ou anti-pneumocoque après vaccination)
")
46
2ème ligne typage lymphocytaire marqueurs T (CD3, CD4, CD8
marqueurs B (CD19, CD20, CD21, Ig) marqueurs NK (CD16) marqueurs d’activation (HLA-DR, CD25)
 marqueurs NK (CD16) marqueurs d’activation (HLA-DR, CD25)")
47
Exploration des déficits immunitaires (II)
2ème ligne évaluation fonctionnelle T (prolifération in vitro, DTH par exemple anatoxine tétanique diluée 1:5) évaluation fonctionnelle B après vaccination, réponse humorale contre antigène protéique (p.ex. tétanos) ou polysaccharidique (pneumocoque) sous-classes IgG (IgG2, IgG3 et IgG4)
 évaluation fonctionnelle B. après vaccination, réponse humorale contre antigène protéique (p.ex. tétanos) ou polysaccharidique (pneumocoque) sous-classes IgG (IgG2, IgG3 et IgG4)")
48
Exploration des déficits immunitaires (III)
2ème ligne complément CH50 (classique et alterne) C3 et C4 Phagocytes phagocytose, réduction du nitrobleu, chimiotactisme
 C3 et C4. Phagocytes. phagocytose, réduction du nitrobleu, chimiotactisme.")
49
Prise en charge générale des patients présentant des déficits immunitaires
mesures générales d’hygiène éliminer allergènes si atopie kiné respiratoire éviter vaccins vivants (polio, fièvre jaune)
")
50
Prise en charge générale des patients présentant des déficits immunitaires
vaccin antigrippal et antipneumococcique antibiothérapies précoces traitements spécifiques immunoglobulines IV pour les déficits en IgG avec répercussion clinique Intérêt des lysats bactériens oraux (bronchovaxom) dans la BPCO?
 dans la BPCO")
51
Infection par le VIH

52
Voir le cours d ’infectiologie (Prof. Demonty
Infection par le VIH aspects épidémiologiques transmission détails des infections opportunistes virologie Voir le cours d ’infectiologie (Prof. Demonty

53
Pathogénie types cellulaires infectés
lymphocytes T CD4+ (y compris thymocytes) lignée macrophagique macrophages cellules dendritiques microglie
 lignée macrophagique. macrophages. cellules dendritiques. microglie.")
54
Interaction Gp120-CD4 Oui mais...

55
Les corécepteurs du VIH

56
Evolution de la lymphocytose CD4

57
Manifestations cliniques
Primoinfection (symptomatique ou non) A : asymptomatique B : signes « mineurs » (type ARC) C : SIDA infections opportunistes (virus, champignons, parasites, bactéries) cancers (Kaposi, LNH, cancer du col, cancer anal) atteinte neurologique (AIDS related dementia)
 A : asymptomatique. B : signes « mineurs » (type ARC) C : SIDA. infections opportunistes (virus, champignons, parasites, bactéries) cancers (Kaposi, LNH, cancer du col, cancer anal) atteinte neurologique (AIDS related dementia)")
58
Manifestations cliniques les plus fréquentes d’entrée en stade C
pneumonie à Pneumocystis Carinii (38%) candidose oesophagienne, trachéale ou bronchique (16%) HIV-associated wasting syndrome (18%) perte de plus de 10% du poids corporel diarrhée (>2selles diarrhéiques /jour pendant un mois) fièvre inexpliquée pendant plus d’un mois
 candidose oesophagienne, trachéale ou bronchique (16%) HIV-associated wasting syndrome (18%) perte de plus de 10% du poids corporel. diarrhée (>2selles diarrhéiques /jour pendant un mois) fièvre inexpliquée pendant plus d’un mois.")
59
HIV et lymphomes Incidence constante malgré HAART
5-10% des patients infectés par le VIH développeront un lymphome 95% des cas : lymphomes dérivés des lymphocytes B

60
Dynamique de l’infection par le VIH
très haute dynamique : ce n’est pas une infection latente fréquence élevée de mutations lors du processus de transcription reverse capacité des souches mutées à résister aux antiviraux (inhibiteurs de RT ou de protéase) persistance d’un sanctuaire rétroviral après des années de traitement efficace
 persistance d’un sanctuaire rétroviral après des années de traitement efficace.")
61
Principes du traitement
Quand traiter? en fonction de symptômes en fonction de paramètres biologiques : essentiellement la lymphocytose CD4 (<350) Comment traiter? au minimum une trithérapie (avec les médicaments actuels) – traitement « à vie »
 Comment traiter au minimum une trithérapie (avec les médicaments actuels) – traitement « à vie »")
62
Principes du traitement
Quel est le but du traitement? à court terme : obtenir un CV indétectable à moyen terme : restaurer une lymphocytose CD4 normale et prévenir les infections opportunistes Quelles sont les causes d ’échec mauvaise compliance patients multitraités dans le passé

63
Principes du traitement
Durée du traitement à vie avec les schémas actuels En évaluation schémas induction/maintenance interruptions programmées

64
Syndrome d’immunoreconstitution
Phénomènes inflammatoires, réactions d’hypersensibilité voire maladies autoimmunitaires pouvant survenir dans les semaines ou les mois qui suivent la mise en place du traitement Rupture de la tolérance aux opportunistes voire au soi

65
Lipodystrophie Effets liés en partie aux traitements
Toxicité mitochondriale Insulinorésistance En partie à la dérégulation immunitaire (TNF-a)
")
66
Suivi du patient séropositif en médecine générale
Conseils de prévention pour l’entourage Soutien psychologique Prophylaxie anti-pneumocystis Accès à un centre spécialisé pour traitement antirétroviral (tous les trois mois) Soutien à la compliance
 Soutien à la compliance.")
67
Diagnostic et suivi de l’infection par le VIH
diagnostic de l’infection par le VIH ELISA et confirmation westernblot (détection d’anticorps) au stade aigu : recherche du génome viral par PCR suivi lymphocytose CD4 ( /mm3) charge virale (nombre de copies de RNA viral par microlitre de plasma) génotype (résistances)
 au stade aigu : recherche du génome viral par PCR. suivi. lymphocytose CD4 ( /mm3) charge virale (nombre de copies de RNA viral par microlitre de plasma) génotype (résistances)")
Présentations similaires