Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
À propos dinhibiteurs et de pharmacologie Au début du XX ième siècle, on ne connaissait que 3 médicaments: -Digitaline (stimulant cardiaque, extrait de la digitale) -Quinine (contre la malaria, extraite dune plante péruvienne, cinchon, amenée en Europe par les jésuites) -Mercure (!) (utilisé contre la syphilis)
 -Quinine (contre la malaria, extraite dune plante péruvienne, cinchon, amenée en Europe par les jésuites) -Mercure (!) (utilisé contre la syphilis)")
2
Comment va agir une drogue -Modification de la fonction dun récepteur dans un organisme ou dans un agent pathogène qui lenvahit. -Ligand ± spécifique pour un enzyme, un canal, une protéine de signalisation. -Très souvent un antagoniste

3
Le développement dune drogue potentielle Celles des dix dernières années sont le résultat du screening dun très grand nombre de produits synthétiques ou naturels Les premiers tests se font in vitro -Liaison à une enzyme impliquée dans la pathologie considérée -Toxicité envers un souche bactérienne donnée -Effet sur des lignées de cellules de mammifères Les tests sur des animaux viennent ultérieurement

4
Molécule candidate avec effet désiré : LEAD COMPOUND Un bon lead: K D < 1 M (minimiser liaisons non spécifiques et assurer un dose thérapeutique faible). Mesures communes de leffet dun drogue: -IC 50 : [ ] produisant 50% inhibition de lenz. -ED 50 : dose qui donne un effet thérapeutique chez 50% des sujets -TD 50 : dose moyenne pour produire un effet toxique -LD 50 : dose léthale pour 50% des sujets

5
IC 50 pour une enzyme Michaélienne TD 50 / ED 50 = Index thérapeutique

6
SARs et QSARs quantitative structure-activity relationships À partir dun lead, on peut tenter le design dun produit plus efficace en lui substituant des groupes methyl- chloro- hydroxyl- benzyl- Pour la plupart des drogues actuelles, de 5 à 10 000 composés dérivés sont synthétisés.

7
La démarche peut suivre une certaine logique. Ainsi, un groupe phényl dun lead qui interagit avec une surface plate dun récepteur donnera un composé à faible affinité si on replace le phényl par un cyclohexyl (non plan) Lidée de base de QSAR repose sur la prémisse quil existe une relation mathématique simple entre lactivité biologique dun produit et ses propriétés physicochimiques. Imaginez que lhydrophobicité soit importante pour obtenir un effet, on sera tenté de changer des substituants pour modifier cette ppté. Lhydrophobicité est généralement formulée par un coefficient de partition, P tel que
 Lidée de base de QSAR repose sur la prémisse quil existe une relation mathématique simple entre lactivité biologique dun produit et ses propriétés physicochimiques. Imaginez que lhydrophobicité soit importante pour obtenir un effet, on sera tenté de changer des substituants pour modifier cette ppté. Lhydrophobicité est généralement formulée par un coefficient de partition, P tel que.")
8
Les constantes (k) sont des indicateurs de la contribution du paramètre considéré à lactivité biologique Lhydrophobicité nest quun paramètre, on peut établir ce genre de relation pour le pKa, le rayon de VdW, lénergie de liaison -H
 sont des indicateurs de la contribution du paramètre considéré à lactivité biologique Lhydrophobicité nest quun paramètre, on peut établir ce genre de relation pour le pKa, le rayon de VdW, lénergie de liaison -H")
9
Approche combinatoire make-many-compounds-ans-see-what-they-do La synthèse parallèle avec 10 variations de R1, R2 et R3 donne 1000 produits différents.

10
La cible du produit final business is the end point La forme commune dadministration est par voie orale. Les barrières à surmonter sont: 1. Résistance aux enzymes digestives et au pH de lestomac ( 1) 2. Absorption du trct. gastrointest. vers circulation sangu. 3. Minimiser la liaison aux protéines du plasma (albumines) et aux tissus adipeux. 4. Résister à la détoxification par le foie 5. Éviter excrétion rénale rapide. 6. Passer facilement des capillaires vers les tissus.
 2. Absorption du trct. gastrointest. vers circulation sangu. 3. Minimiser la liaison aux protéines du plasma (albumines) et aux tissus adipeux. 4. Résister à la détoxification par le foie 5. Éviter excrétion rénale rapide. 6. Passer facilement des capillaires vers les tissus..")
11
7. Traverser la barrière hématoencéphalique le cas échéant (bloque les substances polaires) 8. Atteindre une cible intracellulaire, le cas échéant. La biodisponibilité sera donc une fonction de la dose et de la pharmacocinétique* de la substance considérée. *Décrit interaction avec les barrières citées Évidemment, les barrières 1 et 2 sont contournables par injection.

12
Règles empiriques de Lipinski Un composé aura un faible absorption (perméation) si … 1.P.M. 500 D, question de solubilité et passage au travers des membranes. 2. Possède plus de 5 donneurs de liens –H (groupes NH ou OH) ou plus de 10 accepteurs de liens –H (atomes N ou O), question de polarité. 3. Log P 5 et 6< pK a <8, question de solubilité aqueuse et de passage des formes neutres au travers des membranes.
 ou plus de 10 accepteurs de liens –H (atomes N ou O), question de polarité. 3. Log P 5 et 6< pK a <8, question de solubilité aqueuse et de passage des formes neutres au travers des membranes..")
13
Toxicité et essais cliniques sécurité et efficacité chez humain Phase I. Essais sur 20-100 volontaires sains, sauf si le composé est hautement toxiques (chimiothérapie). Dans ce cas, les volontaires sont des malades souffrant de la maladie cible. Phase II. Essais sur 100-500 malades volontaires. Dose, fréquence, effets secondaires. Tests en simple aveugle contre un placebo, à moins que la maladie soit mortelle. On administre alors le traitement courant au lieu dun placebo. Phase III. Recherche de réactions adverses sur 1000-5000 patients. Tests en double aveugle contre des contrôles.
. Dans ce cas, les volontaires sont des malades souffrant de la maladie cible. Phase II. Essais sur malades volontaires. Dose, fréquence, effets secondaires. Tests en simple aveugle contre un placebo, à moins que la maladie soit mortelle. On administre alors le traitement courant au lieu dun placebo. Phase III. Recherche de réactions adverses sur patients. Tests en double aveugle contre des contrôles..")
14
Le temps et largent Pour 1000 composés testés dans des essais pré- cliniques (~ 3 ans), 1 seul atteint létape des essais cliniques (7-10 ans). Taux de succès: 40% Phase I, 50% Phase II Coût: ~ 300 x 10 6 USD / composé Après la mise en marché, 1 réaction adverse pour 10 000 individus entraine le retrait du produit. (Phase IV)
.")
15
Expl. Le cas de fen-phen (1997)
")
16
Métabolisme des drogues Cytochrome P 450 Une famille de ~100 isoformes de monooxygénases

17
Inhibiteurs des enzymes-clés de HIV-1 … en chiffres -30 millions de décès en 2002 -42 millions de séropositifs -5 millions de nouveaux cas/an

18
1 ière cible: Transcriptase inverse 3-Azido-3-deoxythymidine (AZT; zidovudine)* Problème 1: toxicité pour cell. moelle osseuse oblige à de faibles dosages Problème 2: transcriptase inverse, contrairement aux DNA polymérases ne corrige pas ses erreurs de transcription (~1/10 4 pb) Or, le génome de HIV-1 est d~ 10 000 pb. Ainsi, à un taux de 1 mutation par génome, sous la pression sélective de lAZT, la transcriptase aura tôt fait de développer une résistance. * Synthétisé en 1964 comme agent anticancéreux. Il était inefficace.
 Or, le génome de HIV-1 est d~ pb. Ainsi, à un taux de 1 mutation par génome, sous la pression sélective de lAZT, la transcriptase aura tôt fait de développer une résistance. * Synthétisé en 1964 comme agent anticancéreux. Il était inefficace..")
19
2ième cible: Protéase de Hiv-1 Comme dautres retrovirus, Hiv-1 synthétise des polyprotéines qui ne sont séparées par une protéase, faisant partie elle-même dune polyprotéine, quaprès que le virion soit sorti de la cell. hôte. Sous la forme polyprotéine, le virion est non-infectieux. Cette protéase est donc une cible thérapeutique de choix. Cest une protéase aspartique.

20
Protéases à aspartate (protéases acides) Famille protéolytique avec signature de site actif ASP-THR/SER-GLY Les plus connues: pepsine (digest. stomacale) chymosine(rennine) (digest. veau & bébé h.s.) cathepsines (lysosomiales) renine ( angiotensine I) -secretase (memapsine 2) ( -amyloides)
 chymosine(rennine) (digest. veau & bébé h.s.) cathepsines (lysosomiales) renine ( angiotensine I) -secretase (memapsine 2) ( -amyloides).")
21
Pepsine porcine 326 aa., Les D32 et D215 sont illustrés en spacefill CPK La crevasse centrale peut lier 8 résidus étirés en feuillet

22
Mécanisme

23
Protéase de Hiv-1 Homodimère Les deux D25 sont en spacefill CPK

24
Peptide hypothétique ds site

25
Analogues de lien peptidique (isostères) quon pourrait tester comme inhibiteur de la protéase
 quon pourrait tester comme inhibiteur de la protéase")
26
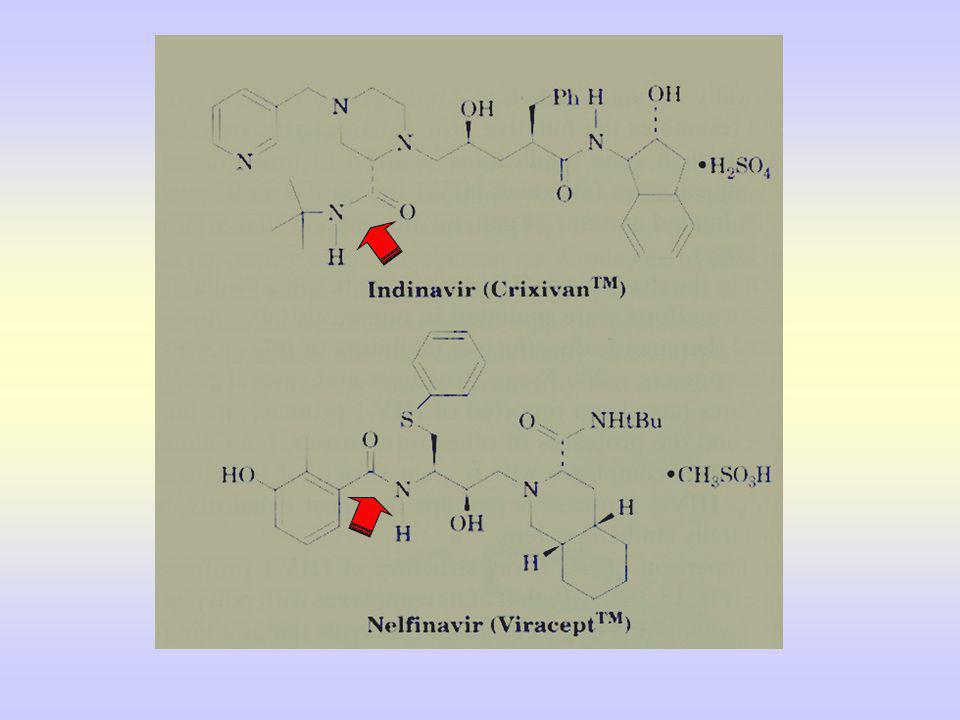
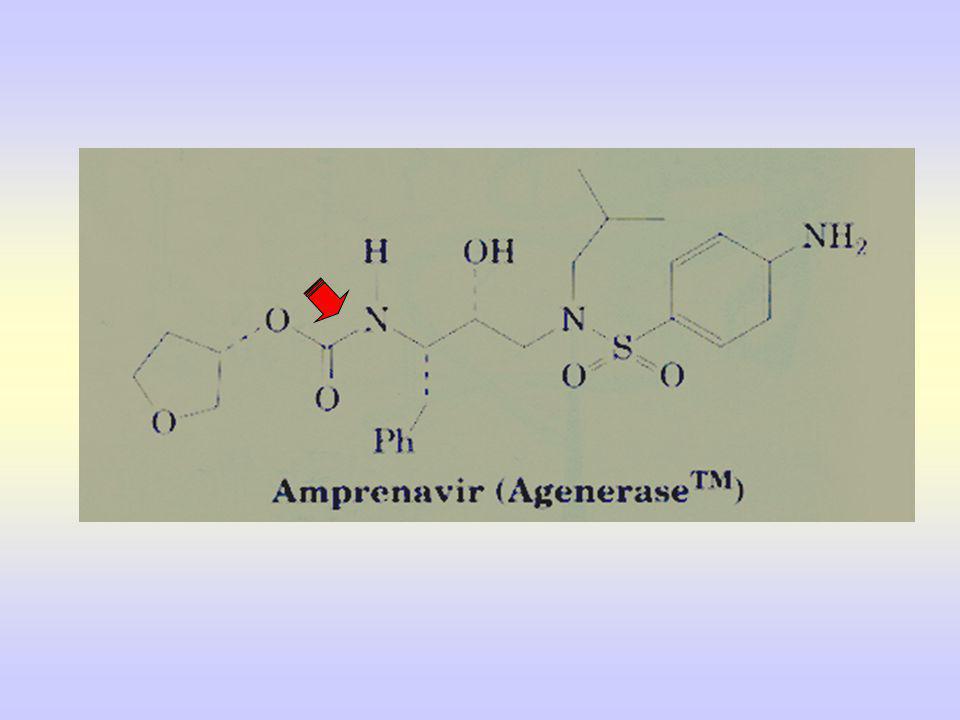
Et les composés peptidomimétiques commercialisés

29
Faute dinhibiteur convenable, on peut se faire fabriquer une enzyme convenable

Présentations similaires












cinétique/pharmaco (toxico)dynamie>")






