Download presentation
1
Drépanocytose Thalassémies Dr Karim Boudjedir (Laboratoire Eurocord) Hôpital Saint Louis IFSI Janvier 2009
 Hôpital Saint Louis IFSI Janvier 2009")
2
Introduction -Hémopathie : pathologie du sang et du tissu lymphoide -bénigne : congénitale ou acquise -maligne : aigue ou chronique . lymphoide ou myéloide -Pronostic variable ,dépend de la morbidité et de la mortalité de chaque pathologie. - Traitement et pronostic sont en fonction de l’hémopathie

3
Définition Hémopathies bénignes congénitales (héréditaire) des globules rouges liée à une anomalie de l’hémoglobine . Font partie d’un groupe hétérogène qu’on appelle : les anémies hémolytiques congénitales (corpusculaires) qu’on classifie en fonction de l’anomalie au niveau du GR : - Les hémoglobinopathies étant les plus fréquentes * anomalies quantitatives de l’HB : Thalassémie (alpha ,béta ) * anomalies qualitatives :Drépanocytose, Hémoglobinose C,…. -Les anomalies de la membrane : la Sphérocytose héréditaire,… -Les déficits enzymatiques : déficit en G6PD, … NB : différentes associations sont possibles ( S – bétathalassémie )
 qu’on classifie en fonction de l’anomalie au niveau du GR : - Les hémoglobinopathies étant les plus fréquentes. * anomalies quantitatives de l’HB : Thalassémie (alpha ,béta ) * anomalies qualitatives :Drépanocytose, Hémoglobinose C,…. -Les anomalies de la membrane : la Sphérocytose héréditaire,… -Les déficits enzymatiques : déficit en G6PD, … NB : différentes associations sont possibles ( S – bétathalassémie )")
5
Drépanocytose

6
Physiopathologie 1 Hémoglobine : role ? constitution ? Hb A2: 22
Globine: 2 chaines et 2 chaines (protéine: assemblage d’AA) Hème (Fe++) Hb A: 22 Hb A2: 22 Hb F: 22 Enfant >6mois et Adulte = 98-99% HbA et 1-2% HbA2 Nouveau-né= HbF +++ Mutation ponctuelle du codon 6 du gène de la globine: par remplacement de l’ac.glutamique par de la valine. l’apparition de l’Hémoglobine S (HbS).
 Hème (Fe++) Hb A: 22. Hb A2: 22. Hb F: 22. Enfant >6mois et Adulte = 98-99% HbA et 1-2% HbA2. Nouveau-né= HbF +++ Mutation ponctuelle du codon 6 du gène de la globine: par remplacement de l’ac.glutamique par de la valine. l’apparition de l’Hémoglobine S (HbS).")
7
Physiopathologie 2 -Dans une situation ordinaire , aucune manifestation clinique. -Dans un contexte de désoxygénation( faible pression d’oxygène), l’ Hb S se polymérise en formation hélicoidales(formation d’un gel ) qui déforment le GR Falciformation des hématies ( forme de faucilles) . - Comme conséquence les GR perdront leur élasticité lors de leurs passages vasculaires (occlusion des petits vaisseaux ). -Facteurs favorisants = Hypoxie Fièvre Acidification sérum définition de la drépanocytose ??
, l’ Hb S se polymérise en formation hélicoidales(formation d’un gel ) qui déforment le GR Falciformation des hématies ( forme de faucilles) . - Comme conséquence les GR perdront leur élasticité ++++ lors de leurs passages vasculaires (occlusion des petits vaisseaux ). -Facteurs favorisants = Hypoxie. Fièvre. Acidification sérum. définition de la drépanocytose")
8
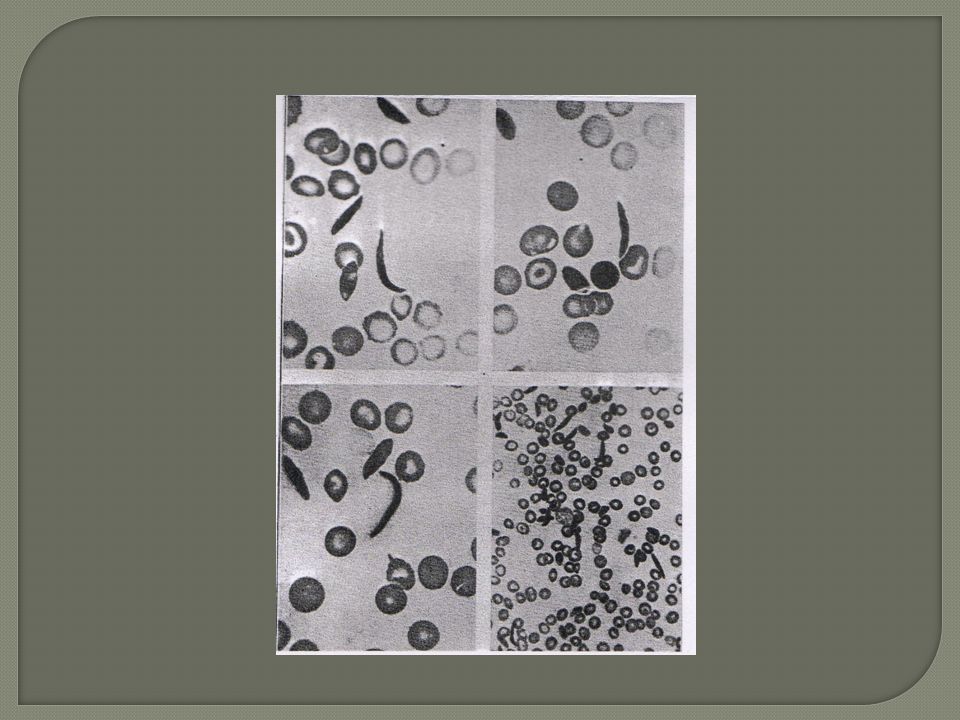
Morphologie Hématies Normal Hb S

10
Schrier, S. ASH Image Bank 2001;2001:100248
Copyright ©2001 American Society of Hematology. Copyright restrictions may apply.

11
Trois conséquences Globule rouge anormal→ Hémolyse (rate ,foie) →Anémie+++ et SPM et/ou HPM Diminution de la déformabilité dans les capillaires →Thromboses vasculaires →douleurs ++++ (localisations variables). La répétition des thromboses →mauvaises irrigations tissulaires →infarcissements (nécroses tissulaires) →dégénérescence et insuffisance de l’organe ++++
. La répétition des thromboses →mauvaises irrigations tissulaires →infarcissements (nécroses tissulaires) →dégénérescence et insuffisance de l’organe ++++")
12
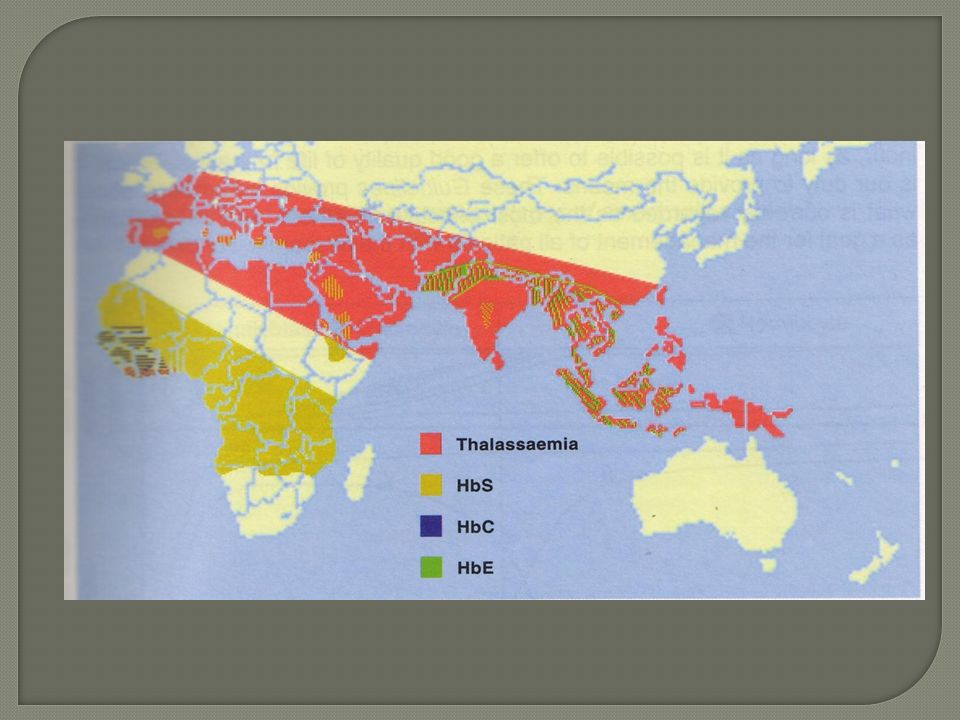
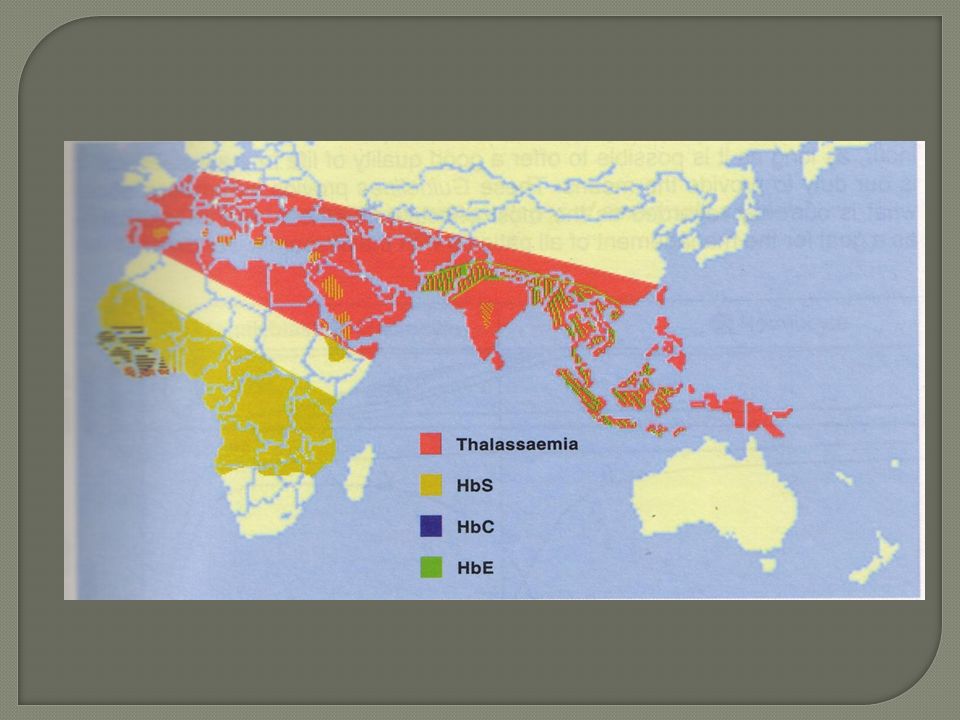
Epidémiologie Maladie génétique , Autosomale récessive (il faut que les 2 parents soient atteints pour être malade). -La + fréquente , incidence :250 à 300 cas /an en France cas d’hétérozygoties . - Population touchées ( Antilles/Guyane) et immigrants d’Afrique noire et pays du Maghreb . Sévérité variable ,notion d’homozygotie ou hétérozygotie . Prévention contre le paludisme.
 et immigrants d’Afrique noire et pays du Maghreb . Sévérité variable ,notion d’homozygotie ou hétérozygotie . Prévention contre le paludisme.")
13
Drépanocytose homozygote (S/S)
1)Etat basal: pas de douleurs Anémie hémolytique modérée : Pâleur +Ictère + splénomégalie (<10 ans) 2)Crises: -Hémolyses aigues (petite enfance) -Crises vaso-occlusives (douleurs) : la petite et grande enfance 3)Infections :(enfance ,adolescence) 4)Complications dégénératives : insuffisance organique (age adulte) ,elles peuvent concernées n’importe quel organe.
Etat basal: pas de douleurs. Anémie hémolytique modérée : Pâleur +Ictère + splénomégalie (<10 ans) 2)Crises: -Hémolyses aigues (petite enfance) -Crises vaso-occlusives (douleurs) : la petite et grande enfance. 3)Infections :(enfance ,adolescence) 4)Complications dégénératives : insuffisance organique (age adulte) ,elles peuvent concernées n’importe quel organe.")
14
Crise Drépanocytaire: clinique
Appelées crises vaso-occlusives Crises douloureuses. Cibles = os, abdomen, thorax (synd thoracique aigu)… Isolée ou multiples. Cas particulier de L’ AVC . Peuvent poser un problème de diagnostic différentiel avec des urgences médicales ou chirurgicales.
… Isolée ou multiples. Cas particulier de L’ AVC . Peuvent poser un problème de diagnostic différentiel avec des urgences médicales ou chirurgicales.")
15
S/S: Biologie -Anémie normochrome , régénérative
-Frottis: Anomalies morphologiques: les drépanocytes. -Electrophorése Hb: HbS de 50 % à 100 % ;HbA dim ; HbA2 =N , Hb F ↑ -Haptoglobine ↓; bilirubine libre ↑ ; fer serique ↑ -Dgc + : électro des 2 parents :drépanocytose hétérozygote (patients asymptomatiques mais porteurs de la maladie) Hb S = 40 % Hb A=60 %
 Hb S = 40 % Hb A=60 %")
16
Prise en charge (1) Support transfusionnel pour le développement harmonieux, quand retentissement important de l’anémie. Education: Eviter circonstances favorisantes (froid, Infections ,déshydratation…) Vaccinations: pneumocoques, Haemophilus, meningocoques) Pénicilline orale ATB à large spectre à la moindre infection.
 Vaccinations: pneumocoques, Haemophilus, meningocoques) Pénicilline orale. ATB à large spectre à la moindre infection.")
17
Prise en charge (2) Hémolyse = Régénération médullaire
Consommation des réserves en folates donc substitution systématique en acide folique. Formes sévères: -Ex-sanguinotransfusion (programme)dans certaines circonstances (grossesse, crises résistantes aux antalgiques ,STA) ou accidents graves (vasculopathies ) - Consultation en urgence voir hospitalisation (indications) (éducation des patients) ++++ -Hydroxyurée (Hydréa°) augmente HbF (indications) -Forme gravissime: Allogreffe de moelle (indications). Conseil génétique ++++
dans certaines circonstances (grossesse, crises résistantes aux antalgiques ,STA) ou accidents graves (vasculopathies ) - Consultation en urgence voir hospitalisation (indications) (éducation des patients) Hydroxyurée (Hydréa°) augmente HbF (indications) -Forme gravissime: Allogreffe de moelle (indications). Conseil génétique ++++")
18
Crise vasooclusive: Traitement
Supprimer facteurs favorisants: froid, hypoxie,… Repos Hyperhydratation Oxygénothérapie Antalgiques éscalades thérapeutiques (paracétamol →morphine à la seringue éléctrique ). Exsanguinotransfusion Trt étiologique (Antibiotiques si Fièvre ,…)
. Exsanguinotransfusion. Trt étiologique (Antibiotiques si Fièvre ,…)")
19
Les règles indispensables pour minimiser la survenue de crises (éducation du patient ).
1/ Bien se laver le corps et les dents pour éviter les microbes provoquant des infections (hygiene de vie ) 2/ Surveiller sa température. 3/ Une température >=38 degrés ! Vite voir un médecin. 4/ Il faut beaucoup boire (environ 3 litres d'eau par jour), 5/ Il faut veiller à ne jamais manquer d'oxygène donc éviter les endroits mal aérés, les hauteurs de plus de 1500 m et les voyages en avion pas ou mal pressurisés 6/ Avoir une bonne alimentation, riche et variée 7/ Surveiller la couleur des yeux et des urines (ictère) 8/ Eviter tout ce qui peut ralentir ou bloquer la circulation du sang : pas de vêtements trop serrés, de positions jambes croisées, etc... 9/ Ne jamais négliger de voir régulièrement le médecin même si tout va bien
 2/ Surveiller sa température. 3/ Une température >=38 degrés ! Vite voir un médecin. 4/ Il faut beaucoup boire (environ 3 litres d eau par jour), 5/ Il faut veiller à ne jamais manquer d oxygène donc éviter les endroits mal aérés, les hauteurs de plus de 1500 m et les voyages en avion pas ou mal pressurisés. 6/ Avoir une bonne alimentation, riche et variée. 7/ Surveiller la couleur des yeux et des urines (ictère) 8/ Eviter tout ce qui peut ralentir ou bloquer la circulation du sang : pas de vêtements trop serrés, de positions jambes croisées, etc... 9/ Ne jamais négliger de voir régulièrement le médecin même si tout va bien.")
20
Evolution/complications :
Anémie chronique: insuffisance cardiaque, les troubles de croissance,.. Thromboses avec Infarctus à répétitions (AVC, ulcère de jambes ,priapisme,STA) puis complications dégénératives (nécroses osseuses, vasculopathies, rétinopathies) et insuffisances multiviscérales (cardiaque,rénale ,…) Infections à répétitions (occlusion des petits vaisseaux spléniques → Asplénie fonctionnelle. Cas particlier de la grossesse et lors d’une anesthésie générale. Pronostic et espérence de vie !! Intéret d’une surveillance annuelle et d’une prise en charge multidisciplinaire
 puis complications dégénératives (nécroses osseuses, vasculopathies, rétinopathies) et insuffisances multiviscérales (cardiaque,rénale ,…) Infections à répétitions (occlusion des petits vaisseaux spléniques → Asplénie fonctionnelle. Cas particlier de la grossesse et lors d’une anesthésie générale. Pronostic et espérence de vie !! Intéret d’une surveillance annuelle et d’une prise en charge multidisciplinaire")
21
Thalassémies

23
Définition A hémolytique congénitale (corpusculaire)
Caractérisée par une diminution de synthése d’une ou plusieurs chaines de globines Thalassémie = Absence d’une ou plusieurs chaines et synthèse d’autre globines en excés Thalassémie = Absence d’une ou plusieurs chaines et synthèse d’autre globines en excés . Maladies génétique, autosomale récessive,de pronostic sévère dans les formes homozygotes .

24
Epidemiologie Fréquence :pourtour méditerranéen, extrême et moyen orient Forme hétérozygote(trait thalassémique) ou homozygote(béta T majeure ou maladie de cooley)
 ou homozygote(béta T majeure ou maladie de cooley)")
26
Béta thalassémie:conséquences
-Hémoglobinisation insuffisante de l’hématie en premier (déficit quantitatif de l’Hb) et presence d’ hématies de petites tailles (hypochromie et microcytose ). - Le déficit en chaine béta va être compenser par la synthese de la chaine gamma (Hb A Hb F) -Production en excés d’Hb F dans le GR hypoxie tissulaire en raison de la forte affinité pour l’O2 activité accrues de la moelle malformations osseuses et retard staturo-ponderal. - Les hématies contenant l’excés de chaines alpha et l’Hb F seront hemolysées dans la MO et en périphérie anémie+ SPM(+/- HPM)+ictère
 et presence d’ hématies de petites tailles (hypochromie et microcytose ). - Le déficit en chaine béta va être compenser par la synthese de la chaine gamma (Hb A Hb F) -Production en excés d’Hb F dans le GR hypoxie tissulaire en raison de la forte affinité pour l’O2 activité accrues de la moelle malformations osseuses et retard staturo-ponderal. - Les hématies contenant l’excés de chaines alpha et l’Hb F seront hemolysées dans la MO et en périphérie anémie+ SPM(+/- HPM)+ictère")
27
Béta thalassémie hétérozygote(1)
France: 1% population générale Aucun symptome clinique Biologie = petits globules rouges (microcytose) mais augmenté en nombre Diagnostic = Electrophorése Hb= Augmentation modéré Hb A2(>3,5%).
 mais augmenté en nombre. Diagnostic = Electrophorése Hb= Augmentation modéré Hb A2(>3,5%).")
28
-Thalassémie hétérozygote (2)
Asymptomatique = Aucun traitement Conseil génétique si le deuxième parent est aussi porteur

29
Béta thalassémie majeure (2 gènes mutés)
Clinique = manifeste à partir du 6 mois Triade d’hémolyse clinique :Paleur +ictère+ SPM Déformations osseuses : faciès mongoloide Retard de croissance (Anémie chronique et hypoxie tissulaire) Anémie chronique = Insuffisance cardiaque, Surcharge en fer (Hémochromatose) Pb endocriniens ,…. NB :Il existe une T homozygote cliniq modérée Biologie : -Anémie < 7gr/dl, microcytaire - Régénérative (la moelle éssaye de compenser le deficit). - Signes d’Hémolyse : bil libre ↑↑ , hapto ↓↓ ,fer sérique ↑↑ -E de L’Hb : Hb F ↑↑↑ Hb A =0 ou ↓↓.(ßß0 ) ou (ßß+) + les 2 parents
 Anémie chronique = Insuffisance cardiaque, Surcharge en fer (Hémochromatose) Pb endocriniens ,…. NB :Il existe une T homozygote cliniq modérée. Biologie : -Anémie < 7gr/dl, microcytaire. - Régénérative (la moelle éssaye de compenser le deficit). - Signes d’Hémolyse : bil libre ↑↑ , hapto ↓↓ ,fer sérique ↑↑ -E de L’Hb : Hb F ↑↑↑ Hb A =0 ou ↓↓.(ßß0 ) ou (ßß+) + les 2 parents.")
30
-Thalassémie homozygote (2)
Sans TRT : évolution vers le décès Traitement basé sur les transfusions Programme de transfusion au long cours (Hb>ou=10g/dl) sang phénotypé ,deleucocyté A folique +pénicilline au long cours+ vaccination. Splénectomie. Pb = surcharge en fer++++ Chélation: Desferal° SC / Exjade° per os++++ Splénectomie++ Maladie grave car développement normal est impossible mortelle avant ans allogreffe de moelle+++ (intrafamilliale 70 % de guérison) Conseil génétique ++++ chez famille à risque et dgc anténatal
 sang phénotypé ,deleucocyté. A folique +pénicilline au long cours+ vaccination. Splénectomie. Pb = surcharge en fer++++ Chélation: Desferal° SC / Exjade° per os++++ Splénectomie++ Maladie grave car développement normal est impossible mortelle avant ans. allogreffe de moelle+++ (intrafamilliale 70 % de guérison) Conseil génétique ++++ chez famille à risque et dgc anténatal.")
31
Alpha Thalassémie Asie Sud-Est, Afrique centrale, Bassin méditerranéen. 1 ou 2 gènes déficitaires : formes silencieuses ou mineures. 3 gènes déficitaires: alpha Thalassémie majeure Révelation précoce (néonatale). Tableau ressemble à la betathalassémie homozygote NB : 4 gènes en moins=Non viable (Mort in utéro).
. Tableau ressemble à la betathalassémie homozygote. NB : 4 gènes en moins=Non viable (Mort in utéro).")