Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
LES ANEMIES HEMOLYTIQUES DE L’ENFANT
Dr.BENCHALLAL

2
OBJECTIF 1. Etablir le diagnostic clinique et biologique d’une anémie hémolytique aigue Et chronique. Etablir le diagnostic clinique et biologique d’une anémie hémolytique congénitale Prendre en charge un enfant présentant une Béta-thalassémie. Prendre en charge un enfant présentant une Drépanocytose. Prendre en charge un enfant présentant un déficit en G6PD.

3
-Elles posent de très difficile problème de prise en charge.
INTRODUCTION -Parmi les anémies hémolytique de l’enfant, les anémies hémolytique congénitales Sont fréquentes dans notre pays en particulier les Thalassémies. -Elles posent de très difficile problème de prise en charge. -Leur prévention n’est malheureusement pas encore faite sur le plan national.

4
-sont diamètre de 8 microns Durée de vie moyenne de 120 jours
Érythrocytes = globules rouges Durée de vie = env. 120 j RAPPEL PHYSIOLOGIQUE 1-LE GLOBULE ROUGE: -l’hématie est une cellule anucléé qui se présente comme un Disque biconcave, plus foncé en périphérie qu’au centre. -sont diamètre de 8 microns Durée de vie moyenne de 120 jours sont hémolyse physiologique siège dans le SRE(MO,foie et rate) ,Principalement la MO. Transport O2 hématies La seule fonction du GR est de véhiculer l’hémoglobine, elle-même transporteuse de l’oxygène des poumons aux tissus. L’important pour l’organisme , c’est la capacité de transport de l’oxygène et donc la concentration d’hémoglobine dans le sang. globules rouges ou hématies, fonction: transport de l'O2
 ,Principalement la MO. Transport O2. hématies. La seule fonction du GR est de véhiculer l’hémoglobine, elle-même transporteuse de l’oxygène des poumons aux tissus. L’important pour l’organisme , c’est la capacité de transport de l’oxygène et donc la concentration d’hémoglobine dans le sang. globules rouges ou hématies, fonction: transport de l O2.")
5
-l’hème :associe un atome de fer et une protoporphyrine.
2-L’HEMOGLOBINE: Constituée par l’association de l’hème(groupe non protéique)et la globine (groupe protéique) -l’hème :associe un atome de fer et une protoporphyrine. -la globine constitue de chaine polypeptidique.
et la globine (groupe protéique) -l’hème :associe un atome de fer et une protoporphyrine. -la globine constitue de chaine polypeptidique.")
6
Profil élécrophorétique de l’hémoglobine
- Chez l’adulte normal: Hb A(alpha2,bété2)=98% Hb A2 (alpha 2,teta2)= 3,3% Hb F(alpha2,gama2)=1% -chez enfant: Hb A= 15 à 40% Hb A2=3,3% Hb F=75% Profil éléctrophorétique de l’adulte est acquis a partir de 6 mois
=98% Hb A2 (alpha 2,teta2)= 3,3% Hb F(alpha2,gama2)=1% -chez enfant: Hb A= 15 à 40% Hb A2=3,3% Hb F=75% Profil éléctrophorétique de l’adulte est acquis. a partir de 6 mois.")
7
Définition et physiopathologie de l’hémolyse
une anémie dite hémolytique si la durée de vie moyenne des GR est raccourcie sans qu’il y ait compensation par une production équivalente d’érythrocytes par la moelle. L’hémolyse entraine une augmentation de la production d’érythropoïétine et de l’activité érythropoitique de la MO l’anémie n’apparait que si la durée de vie est abrégée a un degré tel que les capacités d’une erythropoiése accrue sont débordées. Au dessus de ce seuil l’hemolyse est dite « compensées ». Selon le lieu ou se produit l’hémolyse on distingue l’hémolyse extravasculaire, intra-vasculaire et intra médullaire

8
1- l’hémolyse extravasculaire
Elle s’accomplit sous l’action des macrophage de la MO et surtout de la rate Dans les macrophage,l’Hb est dégradée en un complexe globine-fer-biliverdine Puis le fer et la globine sont détachés, La biliverdine réductase réduit la biliverdine en bilirubine. La bilirubine sera excrétée dans le plasma,sous libre(non conjugée) puis capté par L’hépatocyte ou elle sera conjugée et éliminé par la bile. L’augmentation des réticulocyte,signe d’hyperactivité de la MO. 2- l’hemolyse intra-vasculaire Survient au cours de l’evolution de certaine anémie hémolytique. On note une excés l’hémoglobine plasmatique, colorant le plasma en rouge, Baisse de l’habtoglobine libre Hémoglobinurie et par la suite hémosiderimie. 3-l’hemolyse intra médullaire Elle est exeptionnelle. C’est un avortement des cellules de la lignée erythrocytaite dans la MO Parfois augmentation de la bilirubine et signe d’hyperactivité de la MO.
 puis capté par. L’hépatocyte ou elle sera conjugée et éliminé par la bile. L’augmentation des réticulocyte,signe d’hyperactivité de la MO. 2- l’hemolyse intra-vasculaire. Survient au cours de l’evolution de certaine anémie hémolytique. On note une excés l’hémoglobine plasmatique, colorant le plasma en rouge, Baisse de l’habtoglobine libre. Hémoglobinurie et par la suite hémosiderimie. 3-l’hemolyse intra médullaire. Elle est exeptionnelle. C’est un avortement des cellules de la lignée erythrocytaite dans la MO. Parfois augmentation de la bilirubine et signe d’hyperactivité de la MO.")
9
Classification - la microsphérocytose (maladie de minkowski chauffard.
A. Les anémies par anomalies corpusculaires A.1.Anemies par anomalies corpusculaires hériditaires A.1.a. anomalie de la membrane - la microsphérocytose (maladie de minkowski chauffard. - éllipsocytose et autres. A.1.b.anomalie de structure ou de synthese de l’hémoglobine -les B-thalassémies -hémoglobinopathies hamazygotes (S,C,D,E) -hémoglobinopathies doubles hétérozygotes,(S-C,S-thalassemie) A.2. Les anemies par anomalies corpusculaires acquises -rare chez l’enfant. Tel que l’hémoglobinurie paroxystique nocturne
 -hémoglobinopathies doubles hétérozygotes,(S-C,S-thalassemie) A.2. Les anemies par anomalies corpusculaires acquises. -rare chez l’enfant. Tel que l’hémoglobinurie paroxystique nocturne.")
10
B. Les anemies par anomalies extra corpusculaires
B.1.Les anemies immunologique: -les incompatibilités foeto-maternelles -les anémies hémolytiques autoimmunes -les anémies par allergie médicamenteuse et les transfusions incompatibles B.2.les anemies de cause mécanique -le syndrome hémolytique et urémique -la coagulation intra-vasculaire dissiminée (CIVD) -les protheses valvulaires cardiaques -circulation extracorporelle B.3.les anémies par l’hypersplénismes B.4.les anémies par infectio ou toxique(paludisme,venin de serpent) C. Les anemies par anomalie mixte: Anomalie congénitales sur les quelles se greffe une complication acquise
 -les protheses valvulaires cardiaques. -circulation extracorporelle. B.3.les anémies par l’hypersplénismes. B.4.les anémies par infectio ou toxique(paludisme,venin de serpent) C. Les anemies par anomalie mixte: Anomalie congénitales sur les quelles se greffe une complication acquise.")
11
Clinique 1.Hémolyse chronique: 3 signes sont constant. -la paleur
-l’icter, soit sub-ictere soit foncé, -la splénomégalie, qui est constante 2. Hémolyse aigue ,tableau grave: -paleur intense, dyspnée ,cyanose et parfois oligurie - les urines foncé , - ictere qui fonce progressivement. DIAGNOSTIC POSITIF Clinique

12
Bilan biologique: Il confirme l’hyperhémolyse - Globule rouge diminué,
-Hémoglobine diminué, -réticulocyte augmenté, -bilirubine indirecte augmenté, -fer sérique élevé

13
Diagnostic differentiel
Il faut d’abord : 1.Eliminé les fause anémie, erreur de mesure et hémodilution 2.Discuter les autres anémies: -Anemies post hémorragiques -Hémorragie occulte(anémies carencielles)
")
14
DIAGNOSTIC ETIOLOGIQUE
Les anomalies de l’hémoglobine LES THALASSEMIE Definition Les thalassémies constituent un ensemble hétérogène de maladies génétiques dues à des anomalies des gènes de l’hémoglobine. définies par une diminution de synthèse des chaînes de globine et désignées par la chaîne déficiente : a-thalassémie, b-thalassémie. DIAGNOSTIC ETIOLOGIQUE

15
Classification génétique:
Aspects génétiques: -Le chromosome 16 porte deux gènes a-globine -Chromosome 11 porte les gènes de la famille b-globine. -Les anomalie du génome peuvent aboutir soit a une réduction de synthese de la chaine Béta (B+) soit a une absence de synthése (B-) Classification génétique: -Les B-thalassemies: le déséquilibre dans la production des chaine de globine affecte La chaine B,l’excés de synthese de chaine alpha va avoir des conséquences biologiques et cliniques. Les deux mécanismes principaux a l’origine de l’anemie sont l’hémolyse et l’érythropoiese Énefficace -les alpha thalassemie: L’anomalie moléculaire habituellement responsable des syndromes a-thalassémiques est la délétion [2]. indique le nom du syndrome a-thalassémique résultant de la délétion de un ou de plusieurs gènes a.
 soit a une absence de synthése (B-) Classification génétique: -Les B-thalassemies: le déséquilibre dans la production des chaine de globine affecte La chaine B,l’excés de synthese de chaine alpha va avoir des conséquences biologiques et cliniques. Les deux mécanismes principaux a l’origine de l’anemie sont l’hémolyse et l’érythropoiese Énefficace. -les alpha thalassemie: L’anomalie moléculaire habituellement responsable des syndromes a-thalassémiques est la délétion [2]. indique le nom du syndrome a-thalassémique résultant de la délétion de un ou de plusieurs gènes a.")
16
.Les signes cliniques apparaissent chez l’enfant entre 1 et 5 ans.
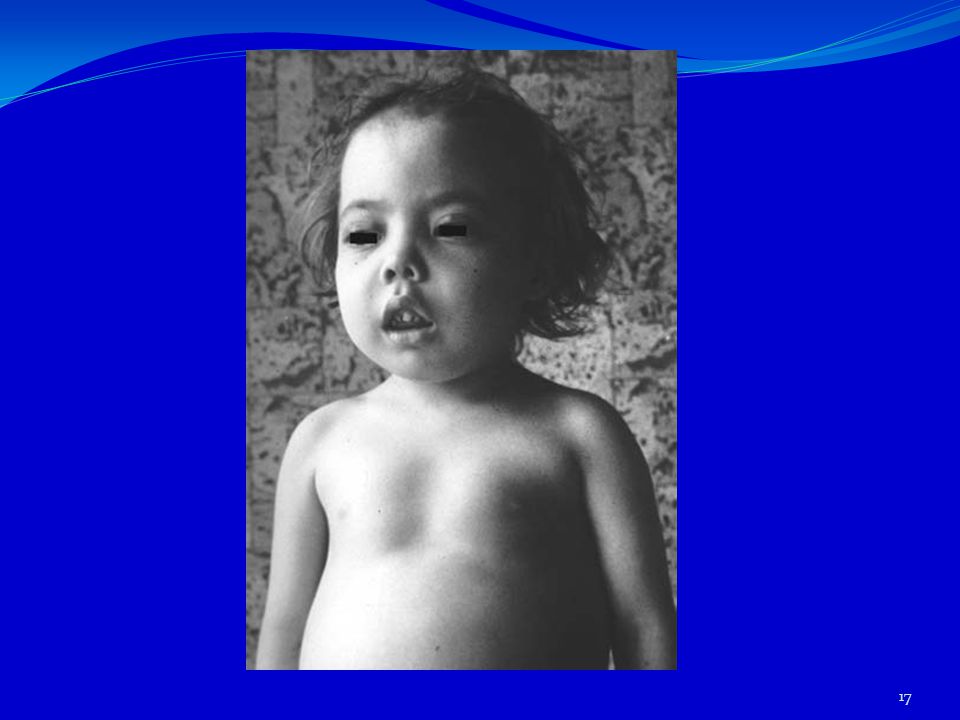
Les B-thalassémies A-Les formes majeur : anémie de COOLEY ASPECT CINIQUE: .Les signes cliniques apparaissent chez l’enfant entre 1 et 5 ans. . le developpement staturo-pondéral est ralenti. .La pâleur est constante, associée fréquemment à un ictère conjonctival dû à l’hémolyse chronique. .Asthénie . Souvent anorexie, .Une hépatosplénomégalie s’installe progressivement , .L’hyperplasie des os plats de la face confère aux enfants un aspect asiatique : les malaires sont élargis, la base du nez aplatie, il existe un hypertélorisme, une protrusion du maxillaire supérieur.donnant un aspect « mongoloide »

18
Signes hématologiques
Signes radiologiques L’hyperplasie de la moelle érythropoïétique épaissit la voûte crânienne, lui donnant un aspect en « poils de brosse ». Elle entraîne aussi une ostéoporose généralisée. Signes hématologiques -Anémie souvent inférieure à 7 g/dl d’hémoglobine, microcytaire,hypochrome --La réticulocytose est voisine de /mm3. -L’examen du frottis sanguin des hématies montre une hypochromie, une anisocytose, des cellule cible,une poïkilocytose et une érythroblastose parfois très élevée, -Le nombre de leucocytes et de plaquettes est souvent normal. L’hyperbilirubinémie traduit l’augmentation de l’hémolyse. L’hypersidérémie et l’hyperferritinémie sont constantes.

19
L’électrophorèse de l’hémoglobine
permet le diagnostic de la b−thalassémie ; le pourcentage d’HbF est constamment augmenté et il persiste (b+-thalassémie) ou pas (b0-thalassémie) de l’HbA
 ou pas (b0-thalassémie) de l’HbA.")
20
L’evolution spontanée de la maladie:
- lée de complication est le risque majeur est l’infection. -La surcharge en fer peut être la cause principal de morbidité et de mortalité.

21
Les formes hétérozygotes de la b-thalassémie
sont généralement asymptomatique du point de vue clinique . leur diagnostic se fait le plus souvent par hasard ou lors d’une enquête familiale Le taux d’Hb est légèrement abaissé. on retrouve une microcyose Elévation du taux de la fraction HB A2.

22
Thalassémie intermédiaire
Ces formes sont caractérisées par une bonne tolérance clinique à l’anémie L’affection est découverte souvent avant 2 ans Les signes constants sont la splénomégalie associé a une paleur une croissance staturo-pondérale normale Anémie souvent modéré, Hb sup ou égale a 7g/dl avec microcytose et hypochromie Il existe une hyper réticulocytose et une dystrophie des GR au frottis sanguin Bilirubine indirect souvent élevé. Les besoins transfusionnels sont modérés ou nuls

23
a-thalassémies Hémoglobinose H
-Défaut de synthése des chaines alpha de l’hémoglobine -L’anomalie moléculaire habituellement responsable des syndromes a-thalassémiques est la délétion. indique le nom du syndrome a-thalassémique résultant de la délétion de un ou de plusieurs gènes a. Hémoglobinose H L’hémoglobinose H résulte de la délétion de trois gènes alpha. Du point de vue clinique: -les patients atteints d’hémoglobinose H ont le plus souvent des formes atténuées de thalassémie. -Parfois, on peut remarquer un faciès de type asiatique, -une splénomégalie -Des anomalies osseuses radiologiques. Du point de vue biologique, l’anémie est en règle générale modérée, entre 9 et 11 g/dl avec microcytose et hypochromie l’électrophorèse de l’hémoglobine montre diminution du taux de A2.

24
L’anasarque foeto-placentaire(Syndrome d’hydrops foetalis
-Ce syndrome correspond à la délétion de quatre gènes a. -Les foetus atteints d’hydrops foetalis décèdent in utero ou dans les minutes qui suivent leur naissance dans un tableau d’anasarque foetoplacentaire. -L’anémie est intense entre 2 et 6 g/dl d’hémoglobine avec la présence d’hémoglobine Bart’s -S’observent chez les couples porteurs chacun d’une a-thalassémie impliquant au moins deux gènes en cis Les trait alpha thalassémiques -Ne comporte aucune manifestation clinique -La survie des érythrocytes marqués au chrome51 normale, une sidérémie normale -Les taux d’hémoglobine A, A2 et F sont normaux.

25
la drépanocytose ,hémoglobinose S:
2. LES HEMOGLOBINOSES -C’est un trouble qualitatif par atteinte da la structure de la globine avec remplacement D’un acide aminé par un autre; -Transmission autosomique recessive Parmis eux on site: la drépanocytose ,hémoglobinose S: -c’est la plus grave et la plus fréquente des hémoglobinose -Atteint surtout les sujets noirs La frequence en algerie est de 0,5 a 3,3%

26
Physiopathologie -la drépanocytose est une maladie génétique de l’hémoglobine due à la mutation du sixième codon de la chaîne b-globine -Entrainant une modification severe de la configuration de Hb -En baisse d’oxygene, se crée une polymérisation moléculaire d’hémoglobine (la falciformation) faisant perdre au hématies leur plasticité normale nécessaires pour passer à travers les petits vaisseaux de l’organisme -le drépanocyte augmente la viscosité du sang expliquant les complications vaso-occlusives de la maladie -les facteurs favorisants: l’hypoxie, l’acidose , la deshydratation ,le froid
 faisant perdre au hématies leur plasticité normale nécessaires pour passer à travers les petits vaisseaux de l’organisme. -le drépanocyte augmente la viscosité du sang expliquant les complications vaso-occlusives de la maladie. -les facteurs favorisants: l’hypoxie, l’acidose , la deshydratation ,le froid.")
27
Génétique -transmise selon le mode récessif autosomique. -Les sujets hétérozygotes sont dits AS et les homozygotes SS

28
Père porteur Non malade Mére porteuse Non malade Enfant non malade Porteur de l’allele mde 2chances sur 4 Enfant malade Un risque sur 4 Enfant non malade Un risque sur4

29
aspects cliniques -l’affection débute entre 5 et 9 mois
-schématiquement la drépanocytose évolue en 3 étapes Avant 5 ans Risque de 3 complications essentilles: syndrome main-pied, tuméfacation répétées aigues et douloureuse des extrémités des mains et des pieds maximum entre 6 et 18 mois les infections severes en raison de l’asplénie fonctionnelle, surtout a pneumocoque la séquestration splénique avec sont risque de choc anémique ;

30
Les accident vaso-occlusifs
Entre 5-15ans Risque de sequestration aigue en raison d’asplénie Crises vaso-occlusives hyperalgiques Le risque infectieux persiste Entre15-20 ans Les accident vaso-occlusifs atteinte cardio-vasculaire(ulcere de jambe,cardiomyopathie) atteinte pulmonaire(infection ,fibrose) atteinte rénales atteinte digestive (lithiase vésiculaire…) atteinte osseuse: épaississement de la voute cranienne déminéralisation des os et des tassement vertebraux
 atteinte pulmonaire(infection ,fibrose) atteinte rénales. atteinte digestive (lithiase vésiculaire…) atteinte osseuse: épaississement de la voute cranienne. déminéralisation des os et des tassement vertebraux.")
31
Les signes hématologique
-L’anémie normocytaire normochrome, régénérative et Hémolytique -hyper bilirubinémie indirecte. -L’hyperleucocytose:constante -Les plaquettes: normales ou légèrement augmentées -Le myelogramme: hyperplasie de la lignée rouge, accessoirement sur les autre lignée L’électrophorèse de l’hémoglobine permet d’affirmer le diagnostic, mettent en évidence la présence d’hémoglobines S, F et A2 ; il n’y a pas d’hémoglobine A

32
Les signes radiologique
-Elargissement des espace médulaire avec amincissement des corticales; -Trabiculations a larges mailles, avec ostéoporose -Aspect en poils de brosse au niveau du crane -Élargissement de l’espace intervertebral

33
Les complications: Les crises vaso-occlusives:
. Résultent de l’obstruction des petit vaisseaux par des hématies falciformes entrainant un infarctus. . souvent provoqué (altitude, froid,acidose,DHA…) . Peut siéger n’importe ou mais surtout au niveau de l’abdomen L’atteinte du squelette: . Tout les os peut etre touché. . le plus frequent , syndrome main-pied, avec oedeme et douleur. . nécroses aseptiques épiphysaires(spécifique de la drépano) . crise douloureuses osseuses,évoquant a tort le RAA
 . Peut siéger n’importe ou mais surtout au niveau de l’abdomen. L’atteinte du squelette: . Tout les os peut etre touché. . le plus frequent , syndrome main-pied, avec oedeme et douleur. . nécroses aseptiques épiphysaires(spécifique de la drépano) . crise douloureuses osseuses,évoquant a tort le RAA.")
34
Les crises aplasiques:
Les crise s de séquestration splénique -menacent le pronostic vital; c’est une urgence thérapeutique -traduisent le piégeage d’une part importante de la masse sanguine dans la rate d’où la chute de l’Hb et risque de collapsus. Les crises aplasiques: -dues à une diminution brusque de l’hyperactivité médullaire, elles se traduisent par une baisse de Hb et des réticulocytes sans majoration de l’ictère -La transfusion est nécessaire jusqu'a retour a l’équilibre. -suspectée devant un enfant anormalement pâle et fatigué. Les crises hyper-hémolytiques - Rares - Elles peuvent être déclenchées par un médicament chez les sujets déficients en G6PD .

35
Les autres complications
Les infections Cause principale de morbidité et de mortalité, les infections graves se voient surtout avant 10 ans Parmi les germes responsable, le pneumocoque plus frequent, puis les salmonelles et l’hémophilus le mycoplasme et les autres germes. Plusieurs tableau peuvent se voire: méningites purulentes ,septicémies , ostéomyélites et pneumopathies aigues Les autres complications Les manifestation neurologique cardio-vasculaire, nephro-urologique ,hépato-biliaire et bronchopulmonaire

36
L’HEMOGLOBINOSE C L’HEMOGLOBINOSE D L’HEMOGLOBINOSE E
-l’acide glutamique en position 6 est remplacé par la lysine, -transmission autosomique récéssive -A l’electrophorese d’Hb: pas de HbA L’HEMOGLOBINOSE D -anémie microcytaire modéré -pas de falciformation, -a l’electrophorese ,l’Hb D migre comme la S. L’HEMOGLOBINOSE E -fréquente en Extreme-Orient, -discrète anémie microcytaire hypochrome

37
4.LES DEFICITS ENZYMATIQUES ERYTHROCYTAIRES
Déficit en G6PD Définition -Anémie hémolytique cong à transmission récessive liée au sexe -Touche les hommes ,les femme sont conductrices - Est due à un déficit en une enzyme érythrocytaire, la G6PD, intervenant dans la glycolyse érythrocytaire - Conséquences : accumulation d’Hb non réduite et fragilisation de la membrane par production de peroxyde. - 2 types : déficit en G6PD de type A = sujets noirs, peu symptomatique (toujours une activité résiduelle de G6PD) de type B = pourtour méditerranéen, Extrême-Orient Déficit en G6PD très répandu dans la planète
 de type B = pourtour méditerranéen, Extrême-Orient. Déficit en G6PD très répandu dans la planète.")
38
Circonstances de découverte
- Découverte 1 à 3 jours après la prise d’un médicament : antipaludéens, sulfamides, quinolones, vitamine C… - Après consommation de fèves (population méditerranéenne), - Suite à une infection virale ; acidose diabétique Clinique Forme habituelle : hémolyse aiguë ++ Après la prise d’un agent déclenchant (médicaments, fèves) crise brutale d’hémolyse intravasculaire o Fièvre, céphalées o Douleurs abdominales et lombaires o Hémoglobinurie (urines rouge sombre) - Dans un 2ème temps : ictère, SMG modérée - Le variant de type B est plus bruyant que le type A
, - Suite à une infection virale ; acidose diabétique. Clinique. Forme habituelle : hémolyse aiguë ++ Après la prise d’un agent déclenchant (médicaments, fèves) crise brutale d’hémolyse intravasculaire. o Fièvre, céphalées. o Douleurs abdominales et lombaires. o Hémoglobinurie (urines rouge sombre) - Dans un 2ème temps : ictère, SMG modérée. - Le variant de type B est plus bruyant que le type A.")
39
Autres formes Forme néo-natale Formes d’hémolyse chronique (rares)
")
40
Diagnostic biologique Hémogramme
- Anémie profonde normochrome, normocytaire, régénérative - Plaquettes : normales ou basse - Leucocytes : normaux ou augmentés éventuellement avec une myélémie discrète. Frottis sanguin - Recherche de sphérocytes, de schizocytes dans les formes sévères - Nombreux corps de Heinz : non visibles en coloration MGG, visibles si utilisation de colorants vitaux

41
Diagnostic de certitude
Biochimie - Bilirubine non conjuguée ! - Haptoglobine diminué - Fer sérique normal ou élevé - Hémoglobinurie dans les cas sévères - Bilan rénal à la recherche d’une insuffisance rénale aiguë Eléments négatifs -Test de Coombs négatif -Electrophorèse de l’Hb = normale -Résistance aux solutés hypotoniques = normale Diagnostic de certitude -Repose sur la MEE du déficit en G6PD (A distance d’une transfusion) - Diagnostic en parallèle par PCR de la mutation
 - Diagnostic en parallèle par PCR de la mutation.")
42
c’est le deuxieme plus frequent type de deficit enzymatique
Deficit en pyruvate kinase c’est le deuxieme plus frequent type de deficit enzymatique Transmission autosomique récéssive, Tableau d’anemie hémolytique chronique severe, Diagnostic repose sur les tests d’auto hémolyse perturbé et le dosage du pyruvate kinase

43
(maladie de minkowski-chauffard)
la sphérocytose hériditaire (maladie de minkowski-chauffard) -Maladie hémolytique a transmission dominante a 75% ,a 25% autosomique récéssive -caractérisé par déformation sphérique des GR qui vont s’auto-hémolysé invitro, corrigé par le glucose, -il y aura une diminution de la déformabilité du GR et sa sequestration dans la rate et sa rapide destruction.. clinique: Ictere d’intensité variable, rarement paleur, splenomégalie, constante, modérée, troubles digestifs. Signe hématologique: anemiegénéralementintermittente,modéré,normochrome, régénérative frottis sanguin, presence de sphérocyte Test de fragilité aux solutions hypotoniques
 -Maladie hémolytique a transmission dominante a 75% ,a 25% autosomique récéssive. -caractérisé par déformation sphérique des GR qui vont s’auto-hémolysé invitro, corrigé par le glucose, -il y aura une diminution de la déformabilité du GR et sa sequestration dans la rate et sa rapide destruction.. clinique: Ictere d’intensité variable, rarement paleur, splenomégalie, constante, modérée, troubles digestifs. Signe hématologique: anemiegénéralementintermittente,modéré,normochrome, régénérative. frottis sanguin, presence de sphérocyte. Test de fragilité aux solutions hypotoniques.")
44
LES ANEMIES HEMOLYTIQUES PAR ANOMALIE EXTRA-CORPUSCULAIRE OU ACQUISE
Biochimie bilirubine indirect élevé haptoglobine diminuée sidérémie normale meilleur pronostic que les autres anémie hémolytique risque d’accident de déglobulisation grave , les complications éloignées :lithiase biliaire, trouble de la croissance et de la pubertaire, Diagnostic microsphérocytose + diminution des la résistance osmotique aux solution hypotonique LES ANEMIES HEMOLYTIQUES PAR ANOMALIE EXTRA-CORPUSCULAIRE OU ACQUISE Le GR est normal, il s’agit d’un mécanisme extérieur, physique, chimique, infectieux, mécanique ou immunologique qui est fréquente

45
Les anemies hémolytique autoimmune (A.H.A.I):
anémie hémolytique régénérative presence d’ac anti GR: diagnostiqué par: test de coombs direct des GR l’elution des anticorp fixés a la surface la recherche des auto-anticorps libres l’evolution soit guérissant en quelque jours ou semaine ou evolution chronique sur plusieurs mois ou année l’etiologie dans les formes aigue: surtout infectieuse: grippe varicelle, MNI. Le syndrome hémolytique et urémique Touche le nourrisson atteinte rénale avec anémie hémolytique et thrombopénie.

46
TRAITEMENT LA B-THALASSEMIE c’est un traitement palliatif
il comporte 3 chapitres: -la transfusion -les chelateur de fer -splenectomie buts du traitement: - maintenir un taux d’Hb entre 10 et 11g/dl pour corriger l’anemie et eviter les déformations osseuse, - lutter contre la surcharge en fer -lutter contre l’hypersplénisme

47
modalité du traitement
la transfusion sanguine qualité de sang: concentré érythrocytaire filtré(déleucocyté, déplaquetté),phénotypé pour le Système ABO,Rh,Kell,Kidd et Duffy quantité du sang: 3ml/kg de culot globulaire pour augmenter l’Hb de 1g/dl en général, la quantité a transfuser est de 15ml/kg toute les 3 semaines Transfusion se déroule en 2 a 3h.
,phénotypé pour le Système ABO,Rh,Kell,Kidd et Duffy. quantité du sang: 3ml/kg de culot globulaire pour augmenter l’Hb de 1g/dl. en général, la quantité a transfuser est de 15ml/kg toute les 3 semaines. Transfusion se déroule en 2 a 3h.")
48
Rythme des transfusions:
Régulièrement toute les 3 a 4 semaines Surveillance des transfusions Chaque enfant doit avoir un carnet de transfusion comportant: Hb pré et post-transfusionnelle, volume transfusé et date de transfusion, référence des sachets des culots globulaire, epreuve de compatibilité transfusionnelle grace a cette surveillance on peut calculer le taux moyen d’Hb annuelle et la consommation de sang annuelle (lm/an), pour dépister la consommation excessive Un excès de consommation correspond a une allo immunisation, soit a un hypersplénisme
, pour dépister la consommation excessive. Un excès de consommation correspond a une allo immunisation, soit a un hypersplénisme.")
49
complication du traitement transfusionnel:
-l’intoxication martiale -Allo-immunisation -infections post-transfusionnelles: syphilis, paludisme, hépatite B et C,CMV,HIV,(intérêt de faire des sérologie régulièrement.

50
Le traitement chélateur du fer:
pour préveunir l’hémosidérose post-transfusionnelle la desferrioxamine (Desferal):chélateur de fer on commence le traitement a un taux de ferritinémie autour de 1000ng/ml, en général après une année de transfusion voie d’administration: S/C en 8-10h, chaque jour a l’aide d’une pompe ,effectué la nuit. Voie IM moins efficace, voie IV réservé a des taux de ferritinémie tres élevé. La défériprone cp 500mg,(75mg/kg/24h) effets secondaires: neutropénie, agranulocytose
:chélateur de fer. on commence le traitement a un taux de ferritinémie autour de 1000ng/ml, en général après une année de transfusion. voie d’administration: S/C en 8-10h, chaque jour a l’aide d’une pompe ,effectué la nuit. Voie IM moins efficace, voie IV réservé a des taux de ferritinémie tres élevé. La défériprone cp 500mg,(75mg/kg/24h) effets secondaires: neutropénie, agranulocytose.")
51
la splénectomie Traitement curatif la thérapie génique
survient chez enfant non ou mal transfusé, doit être totale, précédé de vaccination anti-pneumococcique ,après l’age de 5 ans. la thérapie génique le diagnostic anténatal: par biopsie choriale a la 10 eme semaine de grossesse, permettant le choix d’interuption de grossesse. Traitement curatif c’est la greffe de moelle allogénique , nécessite un donneur sain de la fratrie HLA compatible.

52
LE TRAITEMENT DE LA DREPANOCYTOSE
le buts: prevenir les infection et les traiter soulager la douleur traiter les complications les moyens A. transfusion: pas systematique ,anémie généralement tolérée culots globulaires phénotypé.

53
B. Acide folique: 1 à 2 cp de 5mg, 15 jours par mois ,pour lutter contre la carence C. Antibiotique curative et préventive: pénicilline V: U/kg/j Benzathine pénicilline. D. Paracétamol en cas de fievre. E. Traitement de la douleur: l’hyperhydratation et paracetamol, ibuprofene F. vaccination: anti-pneumococcique, anti-hémophilus influenzae.

54
corticothérapie : 2mg/kg/j ,augmenté a 3mg/kg/j si échec.
TRAITEMENT DES ANEMIES HEMOLYTIQUES AUTOIMMUNES corticothérapie : 2mg/kg/j ,augmenté a 3mg/kg/j si échec. efficacité du traitement: Hb =12g/dl et retic moin de /mm3 décroissance de 0,5mg/kg/j puis un jour sur deux autre thérapeutique: splénéctomie, immunogobuline, serum anti-lymphocytaire, immunosuppresseur.

55
TRAITEMENT DU DEFICIT EN G6PD
transfusion si hemolyse aigue mal tolérée, éviter les féves et certain medicaments nocifs TRAITEMENT DE LA SPHEROCYTOSE le seul traitement est la splénectomie.

Présentations similaires

 Hôpital Saint Louis.>")















 Envahissement médullaire puis systématisé par prolifération de cellules hématopoïétiques malignes ETIOLOGIES 1. Idiopathiques Dans.>")


