Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
La maladie de Hodgkin

2
DEFINITION La maladie de Hodgkin est une forme de cancer du système lymphatique. Elle est anatomo-pathologique : Lymphome malin qui se distingue des autres lymphomes par la présence de grandes cellules à noyau polylobés et multinucléolés (les cellules de Sternberg ou cellules de Reed-Sternberg) ou des ses variantes cytologiques. Le terme anglais est Hodgkin's disease ou Hodgkin's lymphoma
 ou des ses variantes cytologiques. Le terme anglais est Hodgkin s disease ou Hodgkin s lymphoma.")
3
HISTORIQUE Description clinique en 1832 par Thomas Hodgkin qui l'a reconnu comme étant une atteinte primitivement ganglionnaire par opposition aux atteintes ganglionnaires secondaires aux infections ou aux cancers. Les descriptions anatomo-pathologiques datent de 40 ans plus tard (Langhans puis Sternberg et Reed). La maladie de Hodgkin est actuellement incluse dans la classification des syndromes lymphoprolifératifs Ceci explique la tendance actuelle à l'appeler lymphome de Hodgkin.
. La maladie de Hodgkin est actuellement incluse dans la classification des syndromes lymphoprolifératifs. Ceci explique la tendance actuelle à l appeler lymphome de Hodgkin.")
4
EPIDEMIOLOGIE Incidence - Aux USA : nouveaux cas de LNH par an, 7400 nouveaux cas de MH par an - Environ 3 nouveaux cas de MH par an pour habitants. - Larges variations géographiques (0,5 à 4 pour ) - Diminution progressive de l'incidence (- 12 % aux USA entre 1973 et 1991) - Prédominance masculine (ratio de 1,4 : 1) - Quelques cas familiaux
 - Diminution progressive de l incidence (- 12 % aux USA entre 1973 et 1991) - Prédominance masculine (ratio de 1,4 : 1) - Quelques cas familiaux.")
5
Distribution bimodale en fonction de l'âge
- Age médian : 27 ans - Pic pour adolescents et adultes jeunes (15 à 35 ans) - Pic pour sujets plus âgés (> 50 ans) - Cette distribution bimodale tend à s'estomper progressivement
 - Pic pour sujets plus âgés (> 50 ans) - Cette distribution bimodale tend à s estomper progressivement.")
6
Facteurs de risque associés Facteur infectieux
- EBV (Virus d'Epstein-Barr) : Des antécédents de mononucléose infectieuse augmentent le risque d'apparition de 2 à 13 fois. Le génome d'EBV est retrouvé dans 20 à 80 % des cellules de Sternberg avec une fréquence maximale pour les sujets jeunes, les sous-types à cellularité mixte et les cas familiaux. La relation de cause à effet entre EBV et MH reste cependant encore difficile à affirmer formellement. - HHV-6 (Human Herpesvirus-6) : Ce virus a été isolé initialement chez des patients immunodéprimés ou atteints de syndrome lymphoprolifératif. Il est par ailleurs responsable de l'exanthème subit. La relation de cause à effet est incertaine. - VIH (Virus de l'Immunodéficience Humaine) : Le risque de MH est majoré de 5 à 8 fois chez les sujets positifs pour le VIH. Au cours du sida, l'incidence des LNH est supérieure à celle des MH
 : Des antécédents de mononucléose infectieuse augmentent le risque d apparition de 2 à 13 fois. Le génome d EBV est retrouvé dans 20 à 80 % des cellules de Sternberg avec une fréquence maximale pour les sujets jeunes, les sous-types à cellularité mixte et les cas familiaux. La relation de cause à effet entre EBV et MH reste cependant encore difficile à affirmer formellement. - HHV-6 (Human Herpesvirus-6) : Ce virus a été isolé initialement chez des patients immunodéprimés ou atteints de syndrome lymphoprolifératif. Il est par ailleurs responsable de l exanthème subit. La relation de cause à effet est incertaine. - VIH (Virus de l Immunodéficience Humaine) : Le risque de MH est majoré de 5 à 8 fois chez les sujets positifs pour le VIH. Au cours du sida, l incidence des LNH est supérieure à celle des MH.")
7
Facteurs environnementaux
Quelques hypothèses mais aucun argument formel Prédisposition génétique Les facteurs génétiques existent certainement mais sont insuffisants isolément : - Existence de cas familiaux (environ 1 % des cas ) - Risque nettement augmenté mais néanmoins très inconstant (< 5 %) pour les jumeaux homozygotes
 - Risque nettement augmenté mais néanmoins très inconstant (< 5 %) pour les jumeaux homozygotes.")
8
Lymphome de Hodgkin classique divisé en quatre variantes :
· lymphome de Hodgkin classique à sclérose nodulaire : associe une structure nodulaire, de larges bandes de fibrose et un aspect particulier des cellules tumorales dites cellules lacunaires (cytoplasme fragile se rétractant à la fixation) · lymphome de Hodgkin classique à cellularité mixte : les cellules tumorales sont plus nombreuses et la quantité de lymphocytes diminue alors qu'augmentent les polynucléaires neutrophiles et éosinophiles. Il existe une fibrose intercellulaire produite par des fibroblastes hypertrophiques · lymphome de Hodgkin classique à déplétion lymphocytaire : les cellules tumorales sont nombreuses, la fibrose est d'intensité variable · lymphome de Hodgkin classique riche en lymphocytes : les cellules tumorales sont rares sur un fond diffus de cellules lymphocytaires
 · lymphome de Hodgkin classique à cellularité mixte : les cellules tumorales sont plus nombreuses et la quantité de lymphocytes diminue alors qu augmentent les polynucléaires neutrophiles et éosinophiles. Il existe une fibrose intercellulaire produite par des fibroblastes hypertrophiques. · lymphome de Hodgkin classique à déplétion lymphocytaire : les cellules tumorales sont nombreuses, la fibrose est d intensité variable. · lymphome de Hodgkin classique riche en lymphocytes : les cellules tumorales sont rares sur un fond diffus de cellules lymphocytaires.")
9
ASPECTS CLINIQUES Histoire naturelle
L'atteinte initialement ganglionnaire unifocale se propage par voie lymphatique aux territoires adjacents avant de disséminer éventuellement par voie hématogène à des organes non lymphoïdes. Par ordre de fréquence, l'atteinte anatomique primitive est d'abord intrathoracique puis cervicale puis inguino-crurale puis lombaire ou axillaire. A partir des territoires intrathoraciques ou cervicaux, l'extension se fait vers les creux sus -claviculaires et les aisselles ; à partir des territoires sous-diaphragmatiques, elle se fait vers les creux sus-claviculaires principalement gauche en respectant habituellement le médiastin. A partir d'un creux axillaire l'extension se fait vers le creux sus-claviculaire homolatéral. Les lésions viscérales peuvent résulter d'une dissémination hématogène mais aussi d'une extension par contiguïté à partir d'une zone ganglionnaire. Selon le mécanisme d'extension viscérale, le pronostic en est fondamentalement différent (ceci explique les classification en stade IE ou IIE si l'atteinte résulte d'un envahissement de contiguïté).
.")
10
Mode de découverte L'un des aspects les plus fréquents est la découverte d'une adénopathie chez un adulte jeune lors d'un examen médical systématique ou par le patient lui-même. La localisation ganglionnaire la plus fréquente est alors cervicale ou sus-claviculaire, moins souvent axillaire et rarement inguinale. La découverte d'une masse médiastinale antérieure sur un thorax de routine n'est pas inhabituelle. Le deuxième mode de révélation fréquent est l'apparition de symptômes dont aucun n'est spécifique mais dont la persistance ou leur association doit faire évoquer cette hypothèse diagnostique : - asthénie - fièvre surtout vespérale, quelquefois entrecoupée de quelques jours d'apyrexie - hypersudation nocturne - perte de poids (> 10 % du poids dans les 6 derniers mois) - prurit qui peut précéder les autres signes de plusieurs semaines
 - prurit qui peut précéder les autres signes de plusieurs semaines.")
11
signes en rapport avec une masse médiastinale
(douleurs, dyspnée, syndrome cave supérieur) - douleurs dans les sites occupés par les adénopathies en particulier à la phase de début de la maladie ; le déclenchement de ces douleurs par l'ingestion d'alcool est une particularité rare mais utile à connaître La présence de l'un au moins de ces symptômes que sont la fièvre, l'hypersudation et l'amaigrissement définit la notion de signes cliniques d'évolutivité (signes généraux).
 - douleurs dans les sites occupés par les adénopathies en particulier à la phase de début de la maladie ; le déclenchement de ces douleurs par l ingestion d alcool est une particularité rare mais utile à connaître La présence de l un au moins de ces symptômes que sont la fièvre, l hypersudation et l amaigrissement définit la notion de signes cliniques d évolutivité (signes généraux).")
13
Examen clinique L'examen clinique mettra en évidence les localisations de la maladie. Les ganglions lymphatiques restent le site privilégié de l'atteinte. Les adénopathies sont plutôt fermes et peuvent être volumineuses, devenant confluantes et formant de grosses masses tumorales. Les principales aires concernées sont le cou, les creux axillaires, le médiastin antérieur et la région lombo-aortique. La rate est souvent envahie (environ 40 % des cas). L'atteinte médiastinale peut mener à un syndrome cave supérieur. L'extension peut se faire vers la plèvre, le péricarde, le poumon et la paroi thoracique. Rate, poumon, moelle osseuse, foie et os sont les sites extra-ganglionnaires les plus fréquemment envahis.
. L atteinte médiastinale peut mener à un syndrome cave supérieur. L extension peut se faire vers la plèvre, le péricarde, le poumon et la paroi thoracique. Rate, poumon, moelle osseuse, foie et os sont les sites extra-ganglionnaires les plus fréquemment envahis.")
14
BILAN PARACLINIQUE Biologie
- Hémogramme : souvent normal, mais sont possibles hyperleucocytose, anémie, hyperplaquettose, éosinophilie, lymphopénie. Les cytopénies sont rares et sont le plus souvent consécutives à un envahissement médullaire sévère ou plus rarement à des atteintes auto-immunes. - Bilan biologique standard incluant un bilan hépatique et rénal ainsi que le dosage de l'albumine (dont la baisse semble être un facteur pronostic défavorable) tests hépatiques et - Recherche d'un éventuel syndrome inflammatoire (VS augmentée, hyperleucocytose, hyperplaquettose, CRP, hyperfibrinogénémie, hyper, alpha2globulinémie, baisse du fer sérique) - Dosage des LDH dont l'augmentation, plus rare que dans les LNH, est le témoin d'une grande évolutivité. - Une augmentation des phosphatases alcalines oriente si elle est importante vers une atteinte hépatique - Envahissement de la moelle osseuse (rare en phase initiale de la maladie) à rechercher par biopsie systématique et non pas par simple médullogramme - Sérologie HIV
 tests hépatiques et. - Recherche d un éventuel syndrome inflammatoire (VS augmentée, hyperleucocytose, hyperplaquettose, CRP, hyperfibrinogénémie, hyper, alpha2globulinémie, baisse du fer sérique) - Dosage des LDH dont l augmentation, plus rare que dans les LNH, est le témoin d une grande évolutivité. - Une augmentation des phosphatases alcalines oriente si elle est importante vers une atteinte hépatique. - Envahissement de la moelle osseuse (rare en phase initiale de la maladie) à rechercher par biopsie systématique et non pas par simple médullogramme. - Sérologie HIV.")
15
Imagerie - L'exploration du thorax et de l'abdomen par radiographie thoracique standard et scanner thoraco-abdominal est indispensable quelle que soit la présentation clinique initiale. - L'échographie abdominale peut être utile notamment pour un suivi rapproché ou en cas de lésions nodulaires hépatiques. Sa sensibilité est cependant inférieure à celle du scanner pour les adénopathies abdominales et l'échographie ne dispense donc pas du scanner. - L'échographie cervicale est une méthode simple et fiable pour la mesure et le précis des adénopathies à l'étage cervical. Le scanner cervical est également un bon examen pour bien apprécier l'atteinte ganglionnaire cervicale. Un examen ORL spécialisé s'impose lorsque existent des localisations cervicales hautes ou spinales. - La lymphographie bi-pédieuse n'est plus utilisée : examen long, invasif et guère plus informatif que le scanner.

16
- L'intérêt de l'IRM dans la détection de localisations médullaires est controversé :sensibilité de 100 % comparativement à la biopsie mais spécificité insuffisante responsable de faux positifs. - La scintigraphie au gallium ne présente guère d'intérêt dans le cadre du bilan initial mais peut être utile dans le bilan de réévaluation pour distinguer masse médiastinale résiduelle fibreuse non tumorale (ne fixant pas le traceur) et persistance de tissu tumoral actif fixant le gallium. - La tomographie d'émission de positrons (PET scan) après administration de fluorodéoxyglucose marqué au 18F, traceur indiquant une augmentation non spécifique du métabolisme cellulaire, semble être un examen de grande valeur. Sa place se situe idéalement dans le bilan initial et dans le bilan de réévaluation.
 et persistance de tissu tumoral actif fixant le gallium. - La tomographie d émission de positrons (PET scan) après administration de fluorodéoxyglucose marqué au 18F, traceur indiquant une augmentation non spécifique du métabolisme cellulaire, semble être un examen de grande valeur. Sa place se situe idéalement dans le bilan initial et dans le bilan de réévaluation.")
17
Autres investigations
Elles découlent du tableau clinique : - Scintigraphie osseuse si point d'appel clinique - Biopsie hépatique si point d'appel clinique ou biologique - Endoscopies digestives et ponction lombaire ne sont pas utiles (rareté de telles localisations)
")
18
CLASSIFICATION L'extension est classée en 4 stades selon la classification d'Ann Arbor : - Stade I : atteinte d'une seule région ganglionnaire ou d'une seule structure lymphocytaire (rate, thymus, anneau de Waldeyer) - Stade II : atteinte de 2 ou plusieurs régions ganglionnaires situés du même côté du diaphragme. Le médiastin supérieur, moyen et inférieur est considéré comme une seule région mais les hiles pulmonaires sont considérés indépendamment du médiastin. - Stade III : atteinte de ganglion ou de structures lymphocytaires de part et d'autre du diaphragme. Ce stade est subdivisé en stade III1 (atteinte sous diaphragmatique limitée à la rate, aux ganglions du hile splénique, coeliaques ou du tronc porte) et en stade III2 (atteinte des régions lombo-aortiques, iliaques ou mésentériques s'associant ou non à l'envahissement des structures du stade III1). - Stade IV : Atteinte d'un viscère (non contiguë à une atteinte ganglionnaire, voir la définition du "E") ou de plusieurs viscères
 - Stade II : atteinte de 2 ou plusieurs régions ganglionnaires situés du même côté du diaphragme. Le médiastin supérieur, moyen et inférieur est considéré comme une seule région mais les hiles pulmonaires sont considérés indépendamment du médiastin. - Stade III : atteinte de ganglion ou de structures lymphocytaires de part et d autre du diaphragme. Ce stade est subdivisé en stade III1 (atteinte sous diaphragmatique limitée à la rate, aux ganglions du hile splénique, coeliaques ou du tronc porte) et en stade III2 (atteinte des régions lombo-aortiques, iliaques ou mésentériques s associant ou non à l envahissement des structures du stade III1). - Stade IV : Atteinte d un viscère (non contiguë à une atteinte ganglionnaire, voir la définition du E ) ou de plusieurs viscères.")
19
Eléments complémentaires de la classification
· A : absence de signes cliniques d'évolutivité (fièvre, hypersudation ou amaigrissement) · B : présence de signes cliniques d'évolutivité · E : envahissement d'un seul viscère par contiguïté à une atteinte ganglionnaire · X : volumineuse masse tumorale définie par : - une masse ganglionnaire supérieure à 10 cm - ou une masse médiastinale de diamètre égal ou supérieur au tiers du diamètre transverse thoracique au niveau du disque intervertébral D5-D6 sur un cliché thoracique debout. Remarque : la classification a été adaptée aux lymphomes non hodgkiniens avec quelques modifications (voir tableau III du chapitre "Lymphomes non hodgkiniens")
 · B : présence de signes cliniques d évolutivité. · E : envahissement d un seul viscère par contiguïté à une atteinte ganglionnaire. · X : volumineuse masse tumorale définie par : - une masse ganglionnaire supérieure à 10 cm. - ou une masse médiastinale de diamètre égal ou supérieur au tiers du diamètre transverse thoracique au niveau du disque intervertébral D5-D6 sur un cliché thoracique debout. Remarque : la classification a été adaptée aux lymphomes non hodgkiniens avec quelques modifications (voir tableau III du chapitre Lymphomes non hodgkiniens )")
20
Selon une base de données internationale portant sur patients, la répartition est la suivante : - Stade I : 21 % - Stade II : 43 %, - Stade III : 23 % - Stade IV : 13 %. Les symptômes B sont plus fréquents dans les stades avancés : 8 % dans les stades I, 23 % dans les stades II, 37 % dans les stades III, 68 % dans les stades IV (sur 2421 malades de Stanford).
.")
21
PRONOSTIC Survie à 10 ans tous stades confondus (série de Stanford) : - 69 % pour les patients traités entre 1960 et 1968 (survie sans rechute de 51 %) - 84 % pour les patients traités entre 1969 et 1980 (survie sans rechute de 67 %) - 92 % pour les patients traités entre 1981 et 1993 (survie sans rechute de 79 %)
 - 84 % pour les patients traités entre 1969 et 1980 (survie sans rechute de 67 %) - 92 % pour les patients traités entre 1981 et 1993 (survie sans rechute de 79 %)")
22
Les principaux facteurs de mauvais pronostiques sont :
- Stade avancé de la maladie - Présence de signes généraux - Gros volume tumoral - Nombre élevé de viscères atteints dans les stades IV - Atteinte de la moelle osseuse - Age (supérieur à 40, 45 ou 50 ans selon les études) - Sexe masculin - Atteinte inguinale - LDH élevées - Anémie Avec les traitements actuels le type histologique semble peu influencer la survie.
 - Sexe masculin. - Atteinte inguinale. - LDH élevées. - Anémie. Avec les traitements actuels le type histologique semble peu influencer la survie.")
23
LES MOYENS THERAPEUTIQUES
La radiothérapie Historiquement la radiothérapie a été utilisée dès 1902 mais les rechutes étaient nombreuses dans les territoires irradiés et adjacents. Les progrès sont venus de la mise en oeuvre d'irradiations segmentaires traitants en même temps toutes les aires soit au-dessus soit au dessous du diaphragme. En sus-diaphragmatique, l'irradiation en mantelet inclut dans sa forme complète les deux régions cervico-sus-claviculaires, les deux régions axillaires, le médiastin et les hiles pulmonaires. Le mantelet peut être modulé : extension à l'anneau de Waldeyer (atteinte rare dans la MH), suppression du médiastin, inclusion d'un volume pulmonaire adjacent en cas d'envahissement du poumon par contiguïté, protection partielle du médiastin inférieur (cœur et péricarde), ... En sous-diaphragmatique, l'irradiation dite en Y renversé vise la région lombaire, les chaînes iliaques et inguinales. Il est habituel d'y associer une barre splénique concernant la rate et le hile splénique. L'irradiation peut être limitée à la barre verticale du Y et à la loge splénique, épargnant ainsi les ovaires et la moelle hématopoïétique du bassin.
, suppression du médiastin, inclusion d un volume pulmonaire adjacent en cas d envahissement du poumon par contiguïté, protection partielle du médiastin inférieur (cœur et péricarde), ... En sous-diaphragmatique, l irradiation dite en Y renversé vise la région lombaire, les chaînes iliaques et inguinales. Il est habituel d y associer une barre splénique concernant la rate et le hile splénique. L irradiation peut être limitée à la barre verticale du Y et à la loge splénique, épargnant ainsi les ovaires et la moelle hématopoïétique du bassin.")
24
La polychimiothérapie
Le protocole MOPP associant mechloréthamine (Caryolysine), Oncovin, procarbazine (Natulan) et prednisone date de Administré de façon cyclique sur 6 mois, il a permis des taux de rémissions complète de 80 %. La toxicité importante (hématotoxicité, intolérance digestive, stérilité et risque leucémogène) a fait rechercher des alternatives : COPP (remplacement de la Caryolysine par du cyclophosphamide (Endoxan), ABVD (Adriamycine, bléomycine, Velbé, Déticène), alternance MOPP et ABVD ou hybride MOPP/ABV. Les résultats obtenus avec l'ABVD sont égaux ou supérieurs à ceux du MOPP avec une moindre toxicité à long terme. De fait les standards actuels sont l'ABVD, l'alternance MOPPABVD ou l'hybride MOPP/ABV. Le MOPP exclusif est déconseillé. Des protocoles plus intenses, allant jusqu'à des conditionnements lourds avec autogreffe de moelle osseuse, ont été proposés pour les formes de mauvais pronostic et les rechutes.
, Oncovin, procarbazine (Natulan) et prednisone date de Administré de façon cyclique sur 6 mois, il a permis des taux de rémissions complète de 80 %. La toxicité importante (hématotoxicité, intolérance digestive, stérilité et risque leucémogène) a fait rechercher des alternatives : COPP (remplacement de la Caryolysine par du cyclophosphamide (Endoxan), ABVD (Adriamycine, bléomycine, Velbé, Déticène), alternance MOPP et ABVD ou hybride MOPP/ABV. Les résultats obtenus avec l ABVD sont égaux ou supérieurs à ceux du MOPP avec une moindre toxicité à long terme. De fait les standards actuels sont l ABVD, l alternance MOPPABVD ou l hybride MOPP/ABV. Le MOPP exclusif est déconseillé. Des protocoles plus intenses, allant jusqu à des conditionnements lourds avec autogreffe de moelle osseuse, ont été proposés pour les formes de mauvais pronostic et les rechutes.")
25
LES STRATEGIES THERAPEUTIQUES
Stade I et II Formes favorables L'irradiation en mantelet seul est insuffisante (en l'absence de laparotomie exploratrice affirmant formellement la négativité à l'étage abdominale). Par contre, les essais ont montré une équivalence d'efficacité entre irradiation lymphoïde subtotale (sus et sousdiaphragmatique) et l'association radio-chimiothérapie. L'analyse des complications à long terme fera préférer l'association d'un nombre limité de cycles de chimiothérapie (3 cycles) et d'une irradiation limitée en volume (territoires initiaux envahis) et en dose. Formes défavorables (atteinte sous diaphragmatique, signes généraux, grosse masse tumorale) L'association chimiothérapie (3 à 6 cycles) plus radiothérapie segmentaire (sus ou sousdiaphragmatique selon la localisation) est recommandée.
. Par contre, les essais ont montré une équivalence d efficacité entre irradiation lymphoïde subtotale (sus et sousdiaphragmatique) et l association radio-chimiothérapie. L analyse des complications à long terme fera préférer l association d un nombre limité de cycles de chimiothérapie (3 cycles) et d une irradiation limitée en volume (territoires initiaux envahis) et en dose. Formes défavorables (atteinte sous diaphragmatique, signes généraux, grosse masse tumorale) L association chimiothérapie (3 à 6 cycles) plus radiothérapie segmentaire (sus ou sousdiaphragmatique selon la localisation) est recommandée.")
26
Stade III A L'association chimiothérapie (3 à 6 cycles) plus radiothérapie sus et sous-diaphragmatique doit être préférée. Stade III B et IV La chimiothérapie (6 à 8 cures) est l'élément dominant mais la radiothérapie garde une place pour la prévention des rechutes ganglionnaires en particulier si la rémission a été longue à obtenir sous chimiothérapie (plus de 3 cycles) ou s'il s'agissait de grosses masses tumorales. Chez l'enfant La tendance est de limiter la radiothérapie (en indication, en champ et en dose) compte tenu de ses répercussions sur la croissance notamment.
 est l élément dominant mais la radiothérapie garde une place pour la prévention des rechutes ganglionnaires en particulier si la rémission a été longue à obtenir sous chimiothérapie (plus de 3 cycles) ou s il s agissait de grosses masses tumorales. Chez l enfant. La tendance est de limiter la radiothérapie (en indication, en champ et en dose) compte tenu de ses répercussions sur la croissance notamment.")
27
Evaluation de la réponse au traitement
L'obtention d'une rémission complète est l'objectif thérapeutique. La réponse au traitement sera évaluée sur les données cliniques (disparition des signes généraux et des adénopathies) et d'imagerie. En cas d'atteinte initiale très volumineuse médiastinale ou abdominale, la persistance d'une anomalie tomodensitométrique pose le problème de la nature évolutive ou fibreuse de la lésion résiduelle. La scintigraphie au gallium ou mieux le PET scan peuvent alors aider à faire la distinction. En cas de doute persistant, la ponction sous scanner ou un abord chirurgical direct s'impose pour affirmer la rémission complète ou non. Après arrêt thérapeutique la surveillance ultérieure se fera tous les 2 à 3 mois pendant 2 anspuis tous les 4 à 6 mois pendant 5 ans, puis tous les ans compte tenu de complications tardives possibles.
 et d imagerie. En cas d atteinte initiale très volumineuse médiastinale ou abdominale, la persistance d une anomalie tomodensitométrique pose le problème de la nature évolutive ou fibreuse de la lésion résiduelle. La scintigraphie au gallium ou mieux le PET scan peuvent alors aider à faire la distinction. En cas de doute persistant, la ponction sous scanner ou un abord chirurgical direct s impose pour affirmer la rémission complète ou non. Après arrêt thérapeutique la surveillance ultérieure se fera tous les 2 à 3 mois pendant 2 anspuis tous les 4 à 6 mois pendant 5 ans, puis tous les ans compte tenu de complications tardives possibles.")
28
Complications du traitement
Aux effets classiques immédiat de la chimiothérapie et de la radiothérapie s'ajoutent des complications tardives - Infections : il persiste une lymphopénie durant plusieurs années (en particulier après irradiation des aires ganglionnaires) associée à une baisse des fonctions des lymphocytes restant. Une éventuelle splénectomie (ou l'irradiation splénique) est également un grand facteur de risque infectieux (pneumocoque en particulier imposant une vaccination systématique avant l'intervention). - Insuffisance thyroïdienne : d'installation progressive, elle est extrêmement fréquente après irradiation cervicale (incidence cumulée de 50 % à 20 ans dans la série de Stanford). Sa recherche doit être systématique par un dosage de la TSH, une à deux fois par an.
 associée à une baisse des fonctions des lymphocytes restant. Une éventuelle splénectomie (ou l irradiation splénique) est également un grand facteur de risque infectieux (pneumocoque en particulier imposant une vaccination systématique avant l intervention). - Insuffisance thyroïdienne : d installation progressive, elle est extrêmement fréquente après irradiation cervicale (incidence cumulée de 50 % à 20 ans dans la série de Stanford). Sa recherche doit être systématique par un dosage de la TSH, une à deux fois par an.")
29
Complications cardio-vasculaires tardives :
· péricardite post-radique · insuffisance coronarienne pouvant aller jusqu'à un infarctus du myocarde (incidence cumulée d'IDM de 5 % à 15 ans) après radiothérapie médiastinale (action favorisante de l'athérosclérose par la radiothérapie) · myocardiopathie toxique à l'adriamycine rare dans la MH car les doses cumulées restent habituellement modérées.
 après radiothérapie médiastinale (action favorisante de l athérosclérose par la radiothérapie) · myocardiopathie toxique à l adriamycine rare dans la MH car les doses cumulées restent habituellement modérées.")
30
Cancers secondaires : le taux d'incidence cumulée de leucémies et myélodysplasies secondaires peut atteindre 10 % à 10 ans dans certaines séries. Il est plus élevé pour les patients traités par chimiothérapie seule ou par association chimiothérapie + radiothérapie que pour les patients traités par radiothérapie seule. Le risque est dépendant des doses de chimiothérapie administrées et des agents utilisés. Des lymphomes non hodgkiniens et d'autres cancers secondaires (pour lesquels la radiothérapie est certainement en cause : sein, thyroïde, os, mélanome, poumon, ...) ont été observés avec une fréquence accrue.
 ont été observés avec une fréquence accrue.")
31
Troubles de la croissance : consécutifs à une radiothérapie dans le jeune âge, ils peuvent être responsables de séquelles importantes (cyphose, thorax étroit, ...) - Stérilité : les conséquences sur la fonction reproductrice dépendent de l'âge, du sexe, de l'état pubertaire, des cytostatiques et des doses utilisés. Plus de quatre cycles de MOPP administrés avant la puberté entraînent de façon constante une stérilité chez le garçon. Le risque lié à la chimio est moindre chez la femme. La radiothérapie pelvienne augmente le risque en particulier chez la femme. Une congélation de sperme doit être proposé aux adolescents et adultes jeunes. Le rôle protecteur des analogues de la LHRH chez la femme est encore controversé. - fibrose pulmonaire : secondaire à la radiothérapie elle reste limité au champ d'irradiation. Elle apparaît 1 à 6 mois après la radiothérapie et peut être symptomatique (fièvre, toux, dyspnée).
.")
32
LYMPHOMES NON-HODGKINIENS

33
DEFINITION Les lymphomes non hodgkiniens (LNH) représentent un groupe hétérogène de proliférations malignes du système lymphoïde dont le point de départ est extramédullaire. Cette hétérogénéité se traduit par des présentations cliniques, anatomopathologiques, immunologiques et cytogénétiques variées et, de ce fait, par un pronostic très différent d'une forme à l'autre. Le terme anglais est "non-Hodgkin's lymphoma". Les lymphomes font partie des syndromes lymphoprolifératifs au même titre que la leucémie lymphocytaire chronique, la leucémie lymphoblastique aiguë, le myélome multiple et la maladie de Hodgkin
 représentent un groupe hétérogène de proliférations malignes du système lymphoïde dont le point de départ est extramédullaire. Cette hétérogénéité se traduit par des présentations cliniques, anatomopathologiques, immunologiques et cytogénétiques variées et, de ce fait, par un pronostic très différent d une forme à l autre. Le terme anglais est non-Hodgkin s lymphoma . Les lymphomes font partie des syndromes lymphoprolifératifs au même titre que la leucémie lymphocytaire chronique, la leucémie lymphoblastique aiguë, le myélome multiple et la maladie de Hodgkin.")
34
EPIDEMIOLOGIE Incidence - augmente de façon régulière avec l'âge - est plus fréquente chez l'homme que chez la femme (sexe ratio d'environ 2:1) - chez l'enfant les LNH représentent 10 % de l'ensemble des cancers et se situent ainsi en 3ème place derrière les leucémies aiguës et les tumeurs cérébrales. - les LNH les plus agressifs se rencontrent davantage chez l'enfant et l'adulte jeune. - en France, le nombre de nouveaux cas de LNH est estimé entre 3000 et 8000 par an. - l'augmentation de l'incidence constatée depuis quelques années n'est expliquée que partiellement par l'association de certains lymphomes à des états d'immunosuppression sévères (SIDA et transplantation d'organe).
.")
35
Facteurs de risque associés
Facteurs infectieux Virus d'Epstein-Barr (EBV) : Le virus est retrouvé dans 95 % des lymphomes de Burkitt endémiques africains et dans 15 % des formes non endémiques. Il est également associé aux LNH des immunodéprimés (présence dans 50 à 70 % des lymphomes après transplantation d'organe ou au cours du SIDA). Les LNH liés à l'EBV sont le plus souvent des lymphomes B. Il peut être détecté dans le tissu tumoral par immunomarquage par l'anticorps anti LMP-1 (antigène de latence membranaire de l'EBV) ou l'anticorps anti-EBNA-2 (antigène nucléaire de l'EBV). - HTLV-1 (Human T lymphoma/leukemia virus) : Le HTLV-1 est associé à la leucémie/lymphome de l'adulte à cellules T survenant avec prédilection dans le sudouest du Japon, aux Caraïbes, en Afrique noire et en Amérique Centrale. Un à 5 % des sujets séropositifs pour le HLTV-1 développent une leucémie/lymphome de l'adulte à cellules T - VIH (virus de l'immunodéficience humaine) : L'incidence des LNH est augmentée chez les sujets VIH +. Le déficit de l'immunité à médiation cellulaire réduit les capacités d'immunosurveillance des cellules infectées par l'EBV ce qui favorise l'apparition de lymphoproliférations.
 : Le virus est retrouvé dans 95 % des lymphomes de Burkitt endémiques africains et dans 15 % des formes non endémiques. Il est également associé aux LNH des immunodéprimés (présence dans 50 à 70 % des lymphomes après transplantation d organe ou au cours du SIDA). Les LNH liés à l EBV sont le plus souvent des lymphomes B. Il peut être détecté dans le tissu tumoral par immunomarquage par l anticorps anti LMP-1 (antigène de latence membranaire de l EBV) ou l anticorps anti-EBNA-2 (antigène nucléaire de l EBV). - HTLV-1 (Human T lymphoma/leukemia virus) : Le HTLV-1 est associé à la leucémie/lymphome de l adulte à cellules T survenant avec prédilection dans le sudouest du Japon, aux Caraïbes, en Afrique noire et en Amérique Centrale. Un à 5 % des sujets séropositifs pour le HLTV-1 développent une leucémie/lymphome de l adulte à cellules T. - VIH (virus de l immunodéficience humaine) : L incidence des LNH est augmentée chez les sujets VIH +. Le déficit de l immunité à médiation cellulaire réduit les capacités d immunosurveillance des cellules infectées par l EBV ce qui favorise l apparition de lymphoproliférations.")
36
- Hépatite C : Les hépatites chroniques à virus C peuvent se compliquer de cryoglobulinémie et de lymphomes B de faible malignité. Une association avec des lymphomes primitifs du foie a également été suggérée. - HHV-6 (Human Herpes Virus 6) : Le HHV-6 est un virus lymphotrope. Il a été isolé chez des patients porteurs de lymphoproliférations variées mais la relation de cause à effet reste incertaine. - HHV-8 (Human Herpes Virus 8) : Certains lymphomes de présentation clinique très particulière (atteintes des séreuses) ont été associés au HHV-8, le plus souvent au cours du SIDA. Il s'agit probablement d'une co-infection avec l'EBV et le mécanisme reste inconnu. Le HHV-8, encore appelé KSHV (Kaposi sarcoma-associated herpes virus) est également associé au sarcome de Kaposi. - Helicobacter pylori : Le lien entre Helicobacter pylori et lymphome gastrique du MALT (mucosa-associated lymphoid tissue) est maintenant établi au même titre qu'avec la maladie ulcéreuse. La bactérie est détectée dans 90 % des cas de lymphome gastrique du MALT sur coupes tissulaires. En l'absence d'envahissement ganglionnaire, l'antibiothérapie plus oméprazole (inhibiteur de pompe à proton) peut faire régresser le lymphome.
 : Le HHV-6 est un virus lymphotrope. Il a été isolé chez des patients porteurs de lymphoproliférations variées mais la relation de cause à effet reste incertaine. - HHV-8 (Human Herpes Virus 8) : Certains lymphomes de présentation clinique très particulière (atteintes des séreuses) ont été associés au HHV-8, le plus souvent au cours du SIDA. Il s agit probablement d une co-infection avec l EBV et le mécanisme reste inconnu. Le HHV-8, encore appelé KSHV (Kaposi sarcoma-associated herpes virus) est également associé au sarcome de Kaposi. - Helicobacter pylori : Le lien entre Helicobacter pylori et lymphome gastrique du MALT (mucosa-associated lymphoid tissue) est maintenant établi au même titre qu avec la maladie ulcéreuse. La bactérie est détectée dans 90 % des cas de lymphome gastrique du MALT sur coupes tissulaires. En l absence d envahissement ganglionnaire, l antibiothérapie plus oméprazole (inhibiteur de pompe à proton) peut faire régresser le lymphome.")
37
Facteurs immunologiques
Les déficits immunitaires congénitaux ou acquis s'accompagnent d'une incidence accrue de lymphomes : - Déficits congénitaux : ataxie-télangiectasie, syndrome de Wiskott-Aldrich. - Déficits acquis · SIDA : incidence des lymphomes estimée à 5 à 10 % des cas de SIDA. Cette incidence augmente avec la prolongation de la survie. Les lymphomes du SIDA sont caractérisés par leur phénotype B, leur agressivité clinique (les types histologiques les plus fréquents sont les lymphomes de Burkitt et les lymphomes diffus à grandes cellules), leurs localisations extra-ganglionnaires (grande fréquence des lymphomes primitifs du cerveau, atteintes digestives et médullaires). L'apparition d'un lymphome chez un sujet séropositif pour le virus de l'immunodéficience humaine fait passer le patient au stade de SIDA. · Transplantations d'organe : incidence maximale dans les transplantations coeurpoumon et lorsque sont utilisés des protocoles d'immunosuppression forte comme ceux incluant les anticorps monoclonaux anti-lymphocytaires.
, leurs localisations extra-ganglionnaires (grande fréquence des lymphomes primitifs du cerveau, atteintes digestives et médullaires). L apparition d un lymphome chez un sujet séropositif pour le virus de l immunodéficience humaine fait passer le patient au stade de SIDA. · Transplantations d organe : incidence maximale dans les transplantations coeurpoumon et lorsque sont utilisés des protocoles d immunosuppression forte comme ceux incluant les anticorps monoclonaux anti-lymphocytaires.")
38
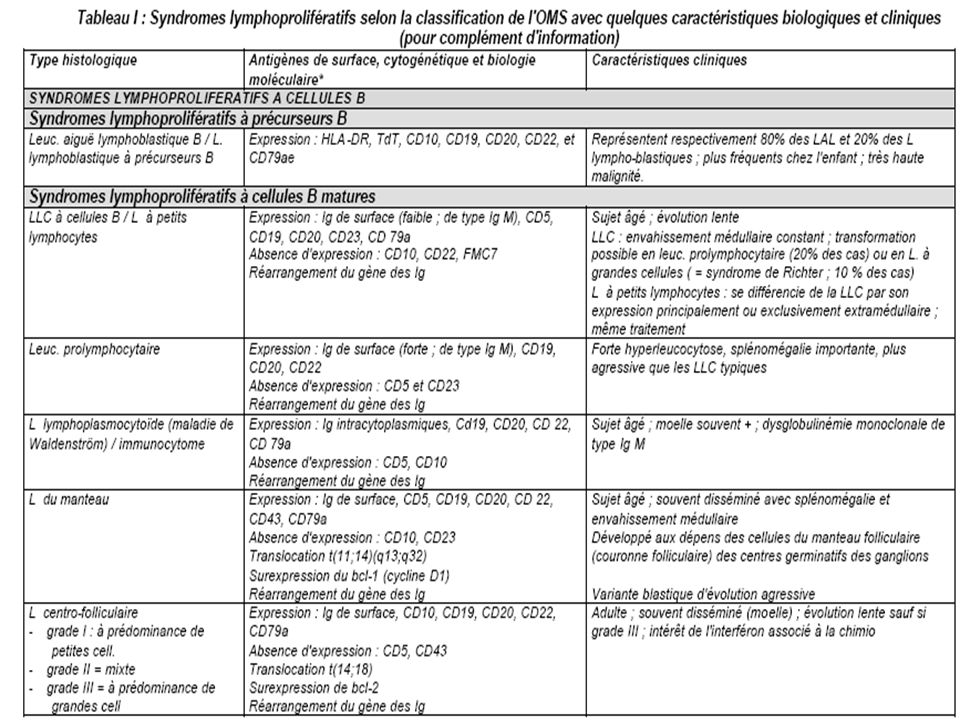
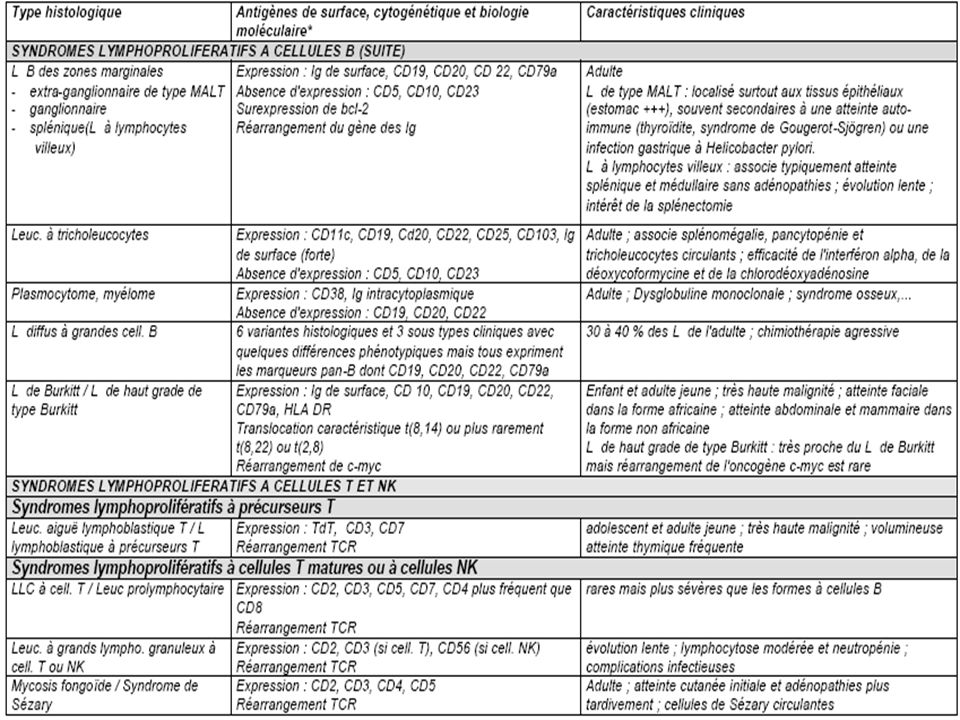
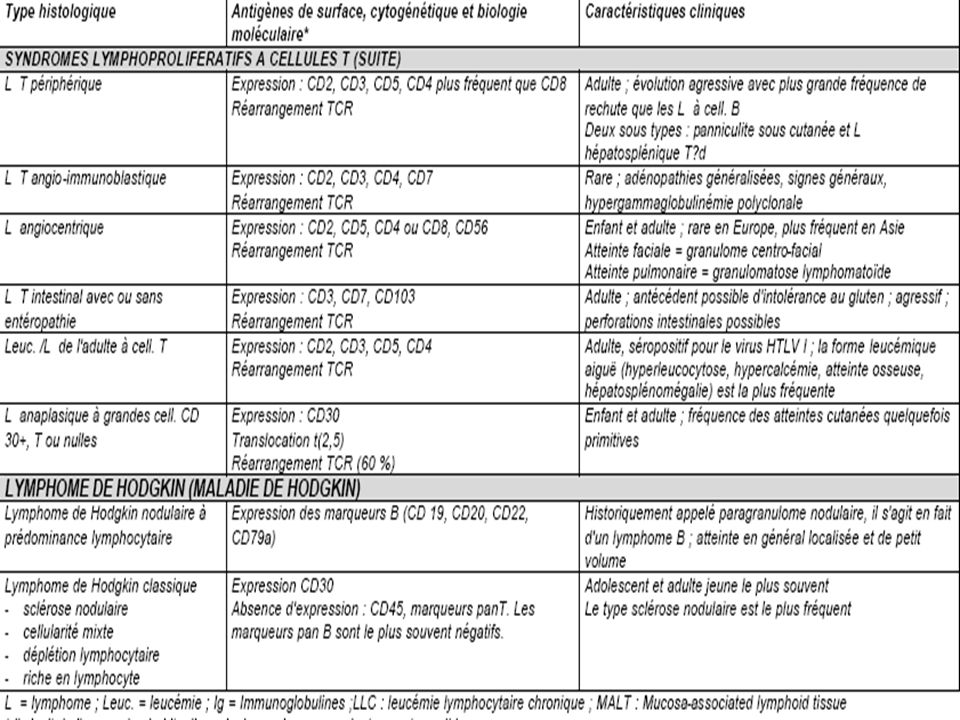
ANATOMIE PATHOLOGIQUE
Histologie La classification actuelle des hémopathies lymphoïdes appelée WHO (World Health Organisation = OMS Organisation Mondiale de la Santé) a été proposée en 1994 et complétée en 1997 (Tableau I). Cette classification est très complexe mais discrimine bien les différentes entités. Elle sépare totalement les syndromes lymphoprolifératifs selon leur phénotype immunologique B ou T et inclut l'ensemble des syndromes lymphoprolifératifs dont plusieurs n'étaient pas pris en compte dans les classifications précédentes (leucémies de la lignée lymphocytaire, leucémie à tricholeucocytes, myélome, maladie de Hodgkin). Elle a également le mérite d'individualiser clairement certaines entités fréquentes mais non prises en compte antérieurement [exemple : lymphome du manteau, lymphomes du MALT (mucosaassociated lymphoid tissue)]. Cette classification repose sur la morphologie mais aussi sur l'immunophénotypage, la cytogénétique et la biologie moléculaire.
 a été proposée en 1994 et complétée en 1997 (Tableau I). Cette classification est très complexe mais discrimine bien les différentes entités. Elle sépare totalement les syndromes lymphoprolifératifs selon leur phénotype immunologique B ou T et inclut l ensemble des syndromes lymphoprolifératifs dont plusieurs n étaient pas pris en compte dans les classifications précédentes (leucémies de la lignée lymphocytaire, leucémie à tricholeucocytes, myélome, maladie de Hodgkin). Elle a également le mérite d individualiser clairement certaines entités fréquentes mais non prises en compte antérieurement [exemple : lymphome du manteau, lymphomes du MALT (mucosaassociated lymphoid tissue)]. Cette classification repose sur la morphologie mais aussi sur l immunophénotypage, la cytogénétique et la biologie moléculaire.")
42
Immunophénotypage C'est un examen essentiel pour le diagnostic précis de la lymphoprolifération. Les leucocytes portent des antigènes de membrane différents selon leur lignée d'origine et leur degré de maturation. La reconnaissance de ces antigènes par l'utilisation d'anticorps monoclonaux permet une identification précise des cellules impliquées. Les anticorps reconnaissant le même antigène sont regroupés en CD (cluster of différenciation = classe de différenciation). Par extension, la définition de CD s'étend à l'antigène leucocytaire reconnu. Il s'agit d'un domaine complexe : plus de 200 CD ont été reconnu, certains étant formés de plusieurs protéines (exemple le CD1 formé de 5 protéines appelées CD1a, CD1b, etc…). Ces antigènes leucocytaires sont des protéines qui ont été regroupés en plusieurs familles (superfamille des immunoglobulines, superfamille des récepteurs aux cytokines, superfamille des intégrines, …) La surexpression des onco-protéines (exemples : bcl-2 dans les lymphomes folliculaires, cycline D1 dans les lymphomes du manteau) peut être mise en évidence en biologie moléculaire ou sur coupes par des anticorps et apporte une aide précieuse au diagnostic.
. Par extension, la définition de CD s étend à l antigène leucocytaire reconnu. Il s agit d un domaine complexe : plus de 200 CD ont été reconnu, certains étant formés de plusieurs protéines (exemple le CD1 formé de 5 protéines appelées CD1a, CD1b, etc…). Ces antigènes leucocytaires sont des protéines qui ont été regroupés en plusieurs familles (superfamille des immunoglobulines, superfamille des récepteurs aux cytokines, superfamille des intégrines, …) La surexpression des onco-protéines (exemples : bcl-2 dans les lymphomes folliculaires, cycline D1 dans les lymphomes du manteau) peut être mise en évidence en biologie moléculaire ou sur coupes par des anticorps et apporte une aide précieuse au diagnostic.")
43
Cytogénétique et biologie moléculaire
Des anomalies cytogénétiques sont retrouvées dans plus de 90% des LNH. Les modifications cytogénétiques impliquent presque toujours un gène des immunoglobulines dans les lymphomes B ou un gène des récepteurs des cellules T dans les lymphomes T. Deux d'entre-elles méritent, de par leur fréquence particulière, d'être développées ici : · Translocation réciproque t (8 ; 14) ou plus rarement t (8 ; 22) ou t (8 ; 2) dans les lymphomes de Burkitt. Ces translocations font voisiner l'oncogène c-myc situé sur le bras long du chromosome 8 et soit le gène des chaînes lourdes d'immunoglobulines (chromosome 14) soit le gène des chaînes légères lambda (chromosome 22) ou kappa (chromosome 2). · Translocation t (14 ; 18) dans plus de 80 % des lymphomes folliculaires à petites cellules clivées mais aussi dans certains cas de lymphomes folliculaires mixtes ou à grandes cellules. Cette translocation juxtapose l'oncogène bcl-2 provenant du chromosome 18 et le gène des chaînes lourdes d'immunoglobulines.
 ou plus rarement t (8 ; 22) ou t (8 ; 2) dans les lymphomes de Burkitt. Ces translocations font voisiner l oncogène c-myc situé sur le bras long du chromosome 8 et soit le gène des chaînes lourdes d immunoglobulines (chromosome 14) soit le gène des chaînes légères lambda (chromosome 22) ou kappa (chromosome 2). · Translocation t (14 ; 18) dans plus de 80 % des lymphomes folliculaires à petites cellules clivées mais aussi dans certains cas de lymphomes folliculaires mixtes ou à grandes cellules. Cette translocation juxtapose l oncogène bcl-2 provenant du chromosome 18 et le gène des chaînes lourdes d immunoglobulines.")
44
L'étude du réarrangement des gènes d'immunoglobulines dans les lymphomes B et des gènes des récepteurs T dans les lymphomes T, techniques réservées à des laboratoiresspécialisés, peut permettre de confirmer le caractère clonal d'une prolifération. La biologie moléculaire peut également détecter des anomalies chromosomiques lorsque les séquences nucléotidiques réarrangées sont bien caractérisées sur le plan moléculaire (translocation t(2 ;5) des lymphomes anaplasiques, translocation t(11 ; 14) des lymphomes du manteau, translocation t(14 ; 18) des lymphomes folliculaires). La biologie moléculaire permet enfin de détecter, après traitement, une maladie résiduelle minime échappant aux méthodes d'investigations morphologiques ou immunologiques conventionnelles.
 des lymphomes anaplasiques, translocation t(11 ; 14) des lymphomes du manteau, translocation t(14 ; 18) des lymphomes folliculaires). La biologie moléculaire permet enfin de détecter, après traitement, une maladie résiduelle minime échappant aux méthodes d investigations morphologiques ou immunologiques conventionnelles.")
45
DIAGNOSTIC ET BILAN D'EXTENSION
Présentation clinique La présentation clinique peut être très variable : - Habituellement le diagnostic est posé suite à la découverte d'adénopathies qui peuvent siéger dans toutes les aires ganglionnaires avec cependant une prédominance au niveau cervical et axillaire. Les adénopathies sont souvent multiples et quelquefois très volumineuses. - Les atteintes extra-ganglionnaires sont fréquentes et peuvent concerner tous les tissus avec une prédilection particulière pour la moelle osseuse, la rate, le tube digestif, la sphère ORL, la peau, le poumon, le foie, la plèvre, l'os, les reins, le système nerveux central. Les localisations préférentielles extra-ganglionnaires sont dépendantes du type histologique du LNH. Ainsi les atteintes médullaires sont particulièrement fréquentes, dès le diagnostic initial, dans les lymphomes B de faible malignité (lymphome à petits lymphocytes, lymphomes folliculaires, lymphome lymphoplasmocytoïde) et dans les lymphome de haute malignité (lymphome lymphoblastique ou lymphome de Burkitt). Les atteintes du système nerveux central se rencontrent plus volontiers dans les lymphomes de haute malignité (lymphome lymphoblastique, lymphome de Burkitt). Les localisations abdominales sont essentiellement associées à des lymphomes B.
 et dans les lymphome de haute malignité (lymphome lymphoblastique ou lymphome de Burkitt). Les atteintes du système nerveux central se rencontrent plus volontiers dans les lymphomes de haute malignité (lymphome lymphoblastique, lymphome de Burkitt). Les localisations abdominales sont essentiellement associées à des lymphomes B.")
46
L'évolutivité de la maladie est également très variable
L'évolutivité de la maladie est également très variable. Ceci a conduit historiquement à une séparation clinique en trois groupes : - les lymphomes de faible malignité (exemples : lymphome à petits lymphocytes, lymphomes folliculaires, lymphome lymphoplasmocytoïde) caractérisés par une évolution lente mais aussi par une moindre sensibilité aux traitements. - les lymphomes de haute malignité (lymphome lymphoblastique ou lymphome de Burkitt) caractérisés par une évolution très agressive avec une forte mortalité précoce mais aussi une grande chimiosensibilité. Les principes thérapeutiques sont identiques à ceux des leucémies aiguës. Il s'agit de réelles urgences thérapeutiques. - les lymphomes de malignité intermédiaires (exemple : lymphome à grandes cellules B) qui se situent entre les deux groupes précédents en terme de génie évolutif. Ils gardent une bonne chimiosensibilité et ont de ce fait le meilleur pronostic à long terme.
 caractérisés par une évolution lente mais aussi par une moindre sensibilité aux traitements. - les lymphomes de haute malignité (lymphome lymphoblastique ou lymphome de Burkitt) caractérisés par une évolution très agressive avec une forte mortalité précoce mais aussi une grande chimiosensibilité. Les principes thérapeutiques sont identiques à ceux des leucémies aiguës. Il s agit de réelles urgences thérapeutiques. - les lymphomes de malignité intermédiaires (exemple : lymphome à grandes cellules B) qui se situent entre les deux groupes précédents en terme de génie évolutif. Ils gardent une bonne chimiosensibilité et ont de ce fait le meilleur pronostic à long terme.")
47
Bilan d'extension Un bilan d'extension complet devra toujours être mené rapidement. Il permet une évaluation exacte de l'ensemble des atteintes et donc une détermination des facteurs pronostiques et un choix raisonné de la stratégie thérapeutique. Ces examens serviront également de référence pour l'appréciation de la réponse au traitement. Le bilan d'extension comprendra au minimum : - Biopsie d'une adénopathie ou d'un tissu envahi (indispensable pour poser le diagnostic). Les investigations devront être complètes et inclure les études morphologiques, immunophénotypique, cytogénétiques et moléculaires. Il est toujours préférable que la biopsie soit faite dans un centre hospitalier capable d'assurer l'ensemble des prestations biologiques.
. Les investigations devront être complètes et inclure les études morphologiques, immunophénotypique, cytogénétiques et moléculaires. Il est toujours préférable que la biopsie soit faite dans un centre hospitalier capable d assurer l ensemble des prestations biologiques.")
48
- Hémogramme à la recherche de signes évoquant une atteinte médullaire (excès de lymphocytes ou cellules lymphoïdes anormales, cytopénie, myélémie) - Biopsie de moelle osseuse (le médullogramme seul est insuffisant) - Phénotypage des lymphocytes et biologie moléculaire au niveau sanguin et médullaire selon le type histologique du lymphome - Radiographie du thorax - Echographie abdominale - Tomodensitométrie thoracique et abdominale - Examen ORL - Ponction lombaire (uniquement lorsque la probabilité d'envahissement est réelle : LNH de malignité intermédiaire et élevée). - Selon le contexte, d'autres examens pourront être utiles : endoscopie digestive, scintigraphie osseuse, biopsie hépatique, ....
 - Phénotypage des lymphocytes et biologie moléculaire au niveau sanguin et médullaire selon le type histologique du lymphome. - Radiographie du thorax. - Echographie abdominale. - Tomodensitométrie thoracique et abdominale. - Examen ORL. - Ponction lombaire (uniquement lorsque la probabilité d envahissement est réelle : LNH de malignité intermédiaire et élevée). - Selon le contexte, d autres examens pourront être utiles : endoscopie digestive, scintigraphie osseuse, biopsie hépatique, ....")
49
D'autres examens compléteront le bilan :
- Bilan hépatique et rénal - Dosage de l'uricémie augmenté dans les formes à prolifération rapide - Dosage de la calcémie pouvant être augmentée en particulier lorsqu'il existe une atteinte osseuse - LDH (lactate deshydrogénases) et bêta-2 microglobuline sériques qui sont le reflet de la prolifération et de la masse tumorale et qui ont une valeur pronostique indépendante des autres facteurs. - Recherche d'une immunoglobuline monoclonale, en particulier dans les lymphomes de faible malignité - Recherche d'une séropositivité pour le VIH, l'hépatite C (et le HTLV-1 dans les LNH T) Evaluation de la fonction cardiaque si le traitement prévu inclut une anthracycline - La tomographie d'émission de positrons (PET) semble être un examen de grande valeur, en particulier pour le suivi de masses résiduelles après traitement. Il est essentiel est de connaître parfaitement l'extension initiale la maladie. De cette connaissance dépendra le choix du traitement et l'appréciation précise de la réponse à ce traitement. La classification en stade la plus utilisée dans les LNH de l'adulte est celled'Ann Arbor développée pour la maladie de Hodgkin (Tableau III).
 et bêta-2 microglobuline sériques qui sont le reflet de la prolifération et de la masse tumorale et qui ont une valeur pronostique indépendante des autres facteurs. - Recherche d une immunoglobuline monoclonale, en particulier dans les lymphomes de faible malignité. - Recherche d une séropositivité pour le VIH, l hépatite C (et le HTLV-1 dans les LNH T) Evaluation de la fonction cardiaque si le traitement prévu inclut une anthracycline. - La tomographie d émission de positrons (PET) semble être un examen de grande valeur, en particulier pour le suivi de masses résiduelles après traitement. Il est essentiel est de connaître parfaitement l extension initiale la maladie. De cette connaissance dépendra le choix du traitement et l appréciation précise de la réponse à ce traitement. La classification en stade la plus utilisée dans les LNH de l adulte est celled Ann Arbor développée pour la maladie de Hodgkin (Tableau III).")
50
FACTEURS PRONOSTIQUES
Les facteurs de mauvais pronostic ont été définis pour les lymphomes nodulaires d'une part et pour les lymphomes de malignité intermédiaire ou élevée d'autre part (Tableau IV). De façon générale, quel que soit le type histologique, l'augmentation des LDH, l'augmentation de la bêta-2 microglobuline, un âge > 60 ans, une forte atteinte de l'état général et une atteinte extra-ganglionnaire diffuse ou multiple sont des facteurs indiscutables de mauvais pronostic
. De façon générale, quel que soit le type histologique, l augmentation des LDH, l augmentation de la bêta-2 microglobuline, un âge > 60 ans, une forte atteinte de l état général et une atteinte extra-ganglionnaire diffuse ou multiple sont des facteurs indiscutables de mauvais pronostic.")
52
PRINCIPES THERAPEUTIQUES
L'approche thérapeutique est fondamentalement différente pour les lymphomes de faible malignité et les lymphomes de malignité intermédiaire et élevée. Lymphomes de faible malignité Les exemples type sont le lymphome à petits lymphocytes, les lymphomes folliculaires ou la maladie de Waldenstrom. Ils sont caractérisés par une évolutivité faible et une médiane de survie longue. Ce sont préférentiellement des lymphomes du sujet âgé. Dix à 40% de ces lymphomes vont se transformer avec le temps (médiane de 4 à 5 ans) en forme histologique plus agressive (perte du caractère folliculaire et/ou augmentation du contingent de grandes cellules). La transformation se caractérise par une progression rapide de la maladie et la médiane de survie est alors inférieure à 1 an.
 en forme histologique plus agressive (perte du caractère folliculaire et/ou augmentation du contingent de grandes cellules). La transformation se caractérise par une progression rapide de la maladie et la médiane de survie est alors inférieure à 1 an.")
53
Selon le stade et l'évolutivité de la maladie le traitement pourra être :
Abstention thérapeutique dans les formes disséminées mais indolentes de lymphomes à petits lymphocytes ou nodulaires à petites cellules clivées. Cette attitude n'est légitime que chez le sujet âgé ou présentant des pathologies intercurrentes pouvant influencer négativement la tolérance à une chimiothérapie. Le traitement chimiothérapique ne sera introduit que lorsque apparaîtront le lymphome franchement. Dans ces conditions la médiane de survie est élevée (supérieure à 10 ans) et le traitement n'est entrepris qu'avec un intervalle de 3 ans chez plus de la moitié des patients. - Radiothérapie des territoires ganglionnaires atteints dans les formes localisées (stade I et II). Ces formes sont peu fréquentes dans les lymphomes de faible malignité et ne concernent que 10 à 20% des patients. Selon les études, la survie en rémission à 10 ans est comprise entre 50 et 80%. L'intérêt de l'adjonction d'une chimiothérapie à la radiothérapie est controversé.
 et le traitement n est entrepris qu avec un intervalle de 3 ans chez plus de la moitié des patients. - Radiothérapie des territoires ganglionnaires atteints dans les formes localisées (stade I et II). Ces formes sont peu fréquentes dans les lymphomes de faible malignité et ne concernent que 10 à 20% des patients. Selon les études, la survie en rémission à 10 ans est comprise entre 50 et 80%. L intérêt de l adjonction d une chimiothérapie à la radiothérapie est controversé.")
54
- Monochimiothérapie orale continue sous forme de chlorambucil (Chloraminophène®)ou de cyclophosphamide (Endoxan®) dans les stades III et IV en l'absence de facteurs de mauvais pronostic. Le taux de rémission complète obtenu est très variable selon les études (de 13 à 65%) mais la médiane de survie reste élevée (voisine de 8 ans). - Polychimiothérapie intraveineuse de type CVP (cyclophosphamide = Endoxan, vincristine = Oncovin, prednisone) ou dérivée du miniCHVP (CVP + doxorubicine = Adriamycine à dose faible) lorsque existent un ou plusieurs facteurs de mauvais pronostic. Le taux de rémission complète est plus élevé qu'avec la monochimiothérapie.
 ou dérivée du miniCHVP (CVP + doxorubicine = Adriamycine à dose faible) lorsque existent un ou plusieurs facteurs de mauvais pronostic. Le taux de rémission complète est plus élevé qu avec la monochimiothérapie.")
55
- Interféron alpha (Roferon® ; Introna®) d'efficacité indiscutable, en association avec la chimiothérapie (miniCHVP), dans les lymphomes folliculaires. Il fait maintenant partie de l'arsenal thérapeutique de première intention dans les formes disséminées avec facteurs de mauvais pronostic. Dans les formes les plus agressives ou lors de rechutes, une chimiothérapie plus lourde éventuellement suivies d'une intensification avec greffe de cellules souches périphériques peut être discutée chez les sujets les plus jeunes. - Une immunothérapie par un anticorps monoclonal anti-CD20 (rituximab) est possible dans les lymphomes folliculaires (qui sont de phénotype B et portent l'antigène CD20). L'efficacité est réelle mais semble plus grande en association avec une chimiothérapie (de type CHOP : cyclophosphamide = Endoxan, doxorubincine = Adriamycine, vincristine = Oncovin, prednisone) Des études sont en cours avec un anticorps anti-CD20 couplé à un isotope radioactif pour renforcer l'action par une irradiation au sein même du tissu lymphomateux.
 est possible dans les lymphomes folliculaires (qui sont de phénotype B et portent l antigène CD20). L efficacité est réelle mais semble plus grande en association avec une chimiothérapie (de type CHOP : cyclophosphamide = Endoxan, doxorubincine = Adriamycine, vincristine = Oncovin, prednisone) Des études sont en cours avec un anticorps anti-CD20 couplé à un isotope radioactif pour renforcer l action par une irradiation au sein même du tissu lymphomateux.")
56
Dans les formes avec plusieurs facteurs de mauvais pronostic, la tendance actuelle est d'intensifier encore la chimiothérapie en particulier en augmentant les posologies des cytostatiques et en maintenant des intercures courts. Ceci n'est possible qu'avec l'utilisation de facteurs de croissance hématopoïétique. Une autre approche est la réalisation précoce, dès la rémission complète obtenue, d'une intensification suivie d'une greffe autologue de cellules souches hématopoïétiques. L'intérêt de ces greffes est validé dans les formes les plus agressives. L'anticorps monoclonal anti-CD20 (rituximab = Mabthera), associé à la chimiothérapie, améliore le taux de réponse et la survie dans les lymphomes B à grandes cellules de l'adulte et représente un progrès majeur dans la prise en charge thérapeutique de ces patients. Le traitement des rechutes est basé sur l'utilisation d'association de drogues non utilisées en première intention (ifosfamide, étoposide, mitoxantrone, cytarabine à haute dose,...). Ces traitements de rattrapage seront complétés par une intensification avec autogreffe de moelle osseuse ou de cellules souches périphériques pour les patients de moins de 60 ans restant chimiosensibles. Les lymphomes lymphoblastiques et les lymphomes de Burkitt sont traités comme des leucémies aiguës.
, associé à la chimiothérapie, améliore le taux de réponse et la survie dans les lymphomes B à grandes cellules de l adulte et représente un progrès majeur dans la prise en charge thérapeutique de ces patients. Le traitement des rechutes est basé sur l utilisation d association de drogues non utilisées en première intention (ifosfamide, étoposide, mitoxantrone, cytarabine à haute dose,...). Ces traitements de rattrapage seront complétés par une intensification avec autogreffe de moelle osseuse ou de cellules souches périphériques pour les patients de moins de 60 ans restant chimiosensibles. Les lymphomes lymphoblastiques et les lymphomes de Burkitt sont traités comme des leucémies aiguës.")
57
Lymphomes de l'enfant Il s'agit pratiquement toujours de formes agressives : lymphomes de Burkitt (35 à 45%), lymphomes lymphoblastiques (25 à 35%), lymphomes diffus à grandes cellules B (15 à 25%). Les traitements sont intenses et soutenus, proches des protocoles de leucémies aiguës de l'enfant et donnent d'excellents résultats avec des rémissions de longue durée dans plus de 70% des cas. Lymphomes au cours du SIDA Leur traitement est également basé sur la chimiothérapie intensive mais il faut tenir compte du risque infectieux accru du fait de l'immunodéficience. Ce risque accru justifie le recours systématique aux facteurs de croissance hématopoïétique. L'anticorps monoclonal anti CD 20 semble intéressant dans les formes B.
, lymphomes lymphoblastiques (25 à 35%), lymphomes diffus à grandes cellules B (15 à 25%). Les traitements sont intenses et soutenus, proches des protocoles de leucémies aiguës de l enfant et donnent d excellents résultats avec des rémissions de longue durée dans plus de 70% des cas. Lymphomes au cours du SIDA. Leur traitement est également basé sur la chimiothérapie intensive mais il faut tenir compte du risque infectieux accru du fait de l immunodéficience. Ce risque accru justifie le recours systématique aux facteurs de croissance hématopoïétique. L anticorps monoclonal anti CD 20 semble intéressant dans les formes B.")
58
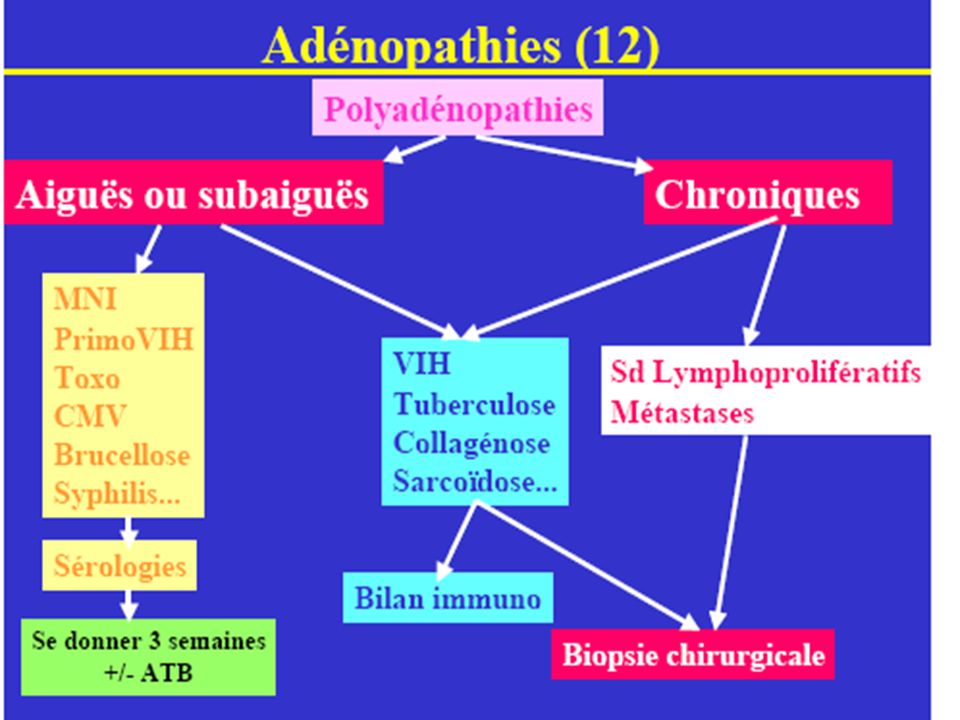
Conduite à tenir devant des adénopathies superficielles

59
Adénopathies (2): Interrogatoire • Age • Activité professionnelle et loisirs • Animaux • FR infection VIH • Séjour en zone d ’endémie parasitaire • ATCD • Traitement et habitus • Signes généraux (AEG, fièvre, sueurs, prurit) • Mode de début , évolution
 • Mode de début , évolution.")
60
Adénopathies (3): Examen clinique • Palpation des aires ganglionnaires : être systématique et efficace! • Caractéristiques des adénopathies: taille, isolées ou groupées, consistance, sensibilité, mobilité, caractère compressif • Examen complet : aires de drainage, foie, rate

61
Adénopathies (4) Territoire de drainage • Aires cervicales : face et cuir chevelu, sphère ORL, thyroïde • Aires susclaviculaires: médiastin, viscères sous diaphragmatiques • Aires axillaires: membres supérieurs, paroi thoraciques, glandes mammaires • Aires inguinales et rétrocrurales : MI, OGE, marge anale

62
DEFINITION : ∗ Adénopathie (ADP) = Hypertrophie pathologique d’un ganglion lymphatique. ∗ Cette hypertrophie est due : − à la prolifération lymphocytaire réactionnelle à une stimulation antigénique soit locorégionale, soit générale. − à l’accumulation de cellules pathologiques filtrées par le ganglion, associée alors à l’hypertrophie lymphoïde réactionnelle. − à la prolifération tumorale primitive du tissu lymphoïde (lymphome) L’architecture normale du ganglion montre des follicules contenant des lymphocytes B, et une « zone T » inter folliculaire. Le ganglion est limité par une capsule. En cas de prolifération tumorale, cette architecture peut être totalement remaniée.
 L’architecture normale du ganglion montre des follicules contenant des lymphocytes B, et une « zone T » inter folliculaire. Le ganglion est limité par une capsule. En cas de prolifération tumorale, cette architecture peut être totalement remaniée.")
63
• SITUATIONS CLINIQUES :
ADP aiguë isolée : satellite d’une pathologie infectieuse + + + PolyADP bénignes : contexte infectieux évocateur (MNI, rubéole, toxoplasmose) Elles peuvent aussi révéler une LAL chez l’enfant ! . ADP isolée chronique sans point d’appel évident : nécessité de la recherche méthodique d’une pathologie dans le territoire de drainage (infection, tumeur). PolyADP chroniques : si bilan infectieux et LLC négatif → Biopsie ganglionnaire chirurgicale ADP profondes : médiastinales (éliminer une sarcoïdose, une tumeur bronchique) ou rétro péritonéales (éliminer une tumeur testiculaire ou gynécologique). Si bilan négatif → Biopsie ganglionnaire chirurgicale
 Elles peuvent aussi révéler une LAL chez l’enfant ! . ADP isolée chronique sans point d’appel évident : nécessité de la recherche méthodique d’une pathologie dans le territoire de drainage (infection, tumeur). PolyADP chroniques : si bilan infectieux et LLC négatif → Biopsie ganglionnaire chirurgicale. ADP profondes : médiastinales (éliminer une sarcoïdose, une tumeur bronchique) ou rétro péritonéales (éliminer une tumeur testiculaire ou gynécologique). Si bilan négatif → Biopsie ganglionnaire chirurgicale.")
64
ETIOLOGIES : ♦ ADP localisées : Cause locorégionale évidente Pas de cause évidente : - bilan topographique orienté - biopsie exérèse ganglionnaire. ♦ ADP disséminées : Cause générale facilement dépistée (MNI, rub, toxo, VIH, Σ, LLC, LA) Pas de cause générale : biopsie ganglionnaire (x1, x2 si nécessaire)
 Pas de cause générale : biopsie ganglionnaire (x1, x2 si nécessaire)")
65
1) HEMOPATHIES MALIGNES Maladie de Hodgkin, LMNH, LLC, Leucémies aigues tumorales, LMC
2) METASTASES GANGLIONNAIRES DE TUMEURS SOLIDES 3) DIVERS ,Infection par le VIH, Tuberculose ganglionnaire, Sarcoïdose, Autres infections (tularémie, brucellose, syphilis).
 METASTASES GANGLIONNAIRES DE TUMEURS SOLIDES. 3) DIVERS ,Infection par le VIH, Tuberculose ganglionnaire, Sarcoïdose, Autres infections (tularémie, brucellose, syphilis).")
66
Adénopathies (5) Questions à se poser • Est ce bien un ganglion lymphatique? • Atteinte localisée ou généralisée? • Pathologie suspectée : maligne ou bénigne? • Atteinte primitive ou secondaire?

67
Adénopathies (6) Diagnostic différentiel • Dans tout les territoires: neurinomes, fibrome, lipome • Territoire cervical: glande salivaire, kyste du tractus thyréoglosse, lymphangiome kystique, kystes branchiaux, dermoïde, grenouillette sushyoïdienne, anévrysme du glomus carotidien, laryngocèle, tumeur thyroïdienne ou musculaire, abcès, côte cervicale. • Territoire axillaire: hidroadénite • Territoire inguinal: hidrosadénite, abcès froid, hernie, kyste du cordon, anévrysme

68
Adénopathies (7) Adénopathies uniques ou groupées • Aiguë +s. Inflammatoires : infection • Cervicale chronique : amygdale,tuberculose • Volumineuse, dure, indolore, fixée, adhérente, infiltrante: malignité+++ • Adénopathie cervicale d ’autant plus suspecte que bas située

69
Adénopathies (8) Polyadénopathies • Contexte aigu et fébrile/ jeune – MNI, VIH, toxo, CMV – Rubéole : occipital++ • + fièvre subaiguë : brucellose, syphilis secondaire, trypanosomiase • Pas de fièvre – VIH, dermatose prurigineuse, maladie systémique (! SGS) – Sd lymphoprolifératifs – Localisation de leucémie myéloïdes – Métastases de carcinome
 – Sd lymphoprolifératifs. – Localisation de leucémie myéloïdes. – Métastases de carcinome.")
70
Polyadénopathies (9) Tableau particulier • SMG: atteinte généralisée : virus, mycobactéries, lupus, sarcoïdose, syndrome lymphoprolifératifs • Adénopathies épitrochléennes : sarcoïdose, syphilis • Occipitale : MNI, rubéole, syphilis

71
Adénopathies (10) Paraclinique • Toute adénopathie > 1-2 mois : biopsie chirurgicale: anapath, bactério • Cytoponction : ne doit pas retarder ou faire surseoir à la biopsie+++ • Bilan minimum si pas de cause locale: – NFS, VS, EPS, CRP – Transa – Sérologies : toxo, VIH, MNI, Syphilis – IDR – RP

74
Unité de Transplantation et Thérapie Cellulaire
Dr Jean EL CHEIKH Unité de Transplantation et Thérapie Cellulaire Département d'Hématologie Institut Paoli-Calmettes

Présentations similaires


















 Envahissement médullaire puis systématisé par prolifération de cellules hématopoïétiques malignes ETIOLOGIES 1. Idiopathiques Dans.>")



