Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Normes et robotique médicale

2
Sommaire Procédures de certification du marquage CE
Directives et Normes européennes de certification Les organismes de certification Les procédures de contrôles et de sécurités Les responsabilités civiles

3
Sommaire Procédures de certification du marquage CE
Directives et Normes européennes de certification Les organismes de certification Les procédures de contrôles et de sécurités Les responsabilités civiles

4
Définition du marquage CE
Inscription apposée de façon visible sur un équipement mis sur le marché européen, soit directement, soit sur une fiche signalétique. Il est apposé par le fabriquant ou le distributeur qui garantit ainsi que l’équipement est conforme aux exigences de la directive européenne associé au produit. Un équipement ainsi marqué peut librement circuler dans toute l’Union Européenne. Les équipements pour lesquels il est prévu une vérification de conformité, le marquage CE est suivi du numéro d’identification de l’organisme notifié.

5
Définition d’un dispositif médical
On entend par dispositif médical tout instrument, appareil, équipement, matière ou autre article, utilisé seul ou en association, y compris le logiciel nécessaire pour le bon fonctionnement de celui-ci, destiné par le fabriquant à être utilisé chez l’homme à des fins médicales et dont l’action principale voulue, dans ou sur le corps humain, n’est pas obtenue par des moyens pharmacologiques ou immunologiques ni par métabolisme, mais dont la fonction peut être assistée par de tel moyens. Ces dispositifs sont destinés à être utilisés à des fins : De diagnostic, de prévention, de contrôle, de traitement ou d’atténuation de la maladie D’étude, de remplacement ou de modification de l’anatomie ou d’un processus physiologique De la maîtrise de la conception.

6
Classification des DM La directive 93/42/CEE regroupe les dispositifs médicaux en 4 classes selon des critères de risque qui peuvent être : la durée d’utilisation, le caractère invasif ou non, le caractère implantable ou non, le type chirurgical ou non CLASSE I faible degré de risque CLASSE IIa moyen degré de risque CLASSE IIb potentiel élevé de risque CLASSE III critiques en matière de risque ( plus particulièrement les dispositifs médicaux implantables actifs) Les robots utilisés à des fins médicales feront partis des classes IIa et IIb
 Les robots utilisés à des fins médicales feront partis des classes IIa et IIb.")
7
Procédures de certification
La certification implique une démarche en 4 étapes : 1. Définir la classe dans laquelle se trouve le dispositif médical concerné. 2. Suivre les modules de preuve ( ou procédure d’évaluation de la conformité) 3. Obtenir et établir les documents attestant de la conformité 4. Apposer le marquage CE
 3. Obtenir et établir les documents attestant de la conformité. 4. Apposer le marquage CE.")
8
Modules de preuves Classe du dispositif
Modules applicables à chaque classe Classe IIa Système complet d’AQ Déclaration CE de conformité Classe IIb Examen CE de Type

9
Sommaire Procédures de certification du marquage CE
Directives et Normes européennes de certification Les organismes de certification Les procédures de contrôles et de sécurités Les responsabilités civiles

10
Normes Européennes Harmonisées
Un fabricant utilisera de préférence les normes européennes harmonisées qui existent pour le dispositif médical ciblé. Elles constituent une sorte de cahier des charges techniques, référence de l’art le plus largement utilisé par les professionnels. Elles ont 2 objectifs : Traduire les exigences essentielles de santé et de sécurité de la directive en spécifications techniques précises. Conférer au dispositifs médicaux une présomption de conformité à la directive. Les deux grosses normes européennes harmonisées applicables aux dispositifs médicaux sont : les normes EN et la série des EN EN normes relatives aux exigences des systèmes qualités EN exigences spécifiques complémentaires aux exigences générales des normes EN pour les fabricant de tous les dispositifs médicaux.

11
Sommaire Procédures de certification du marquage CE
Directives et Normes européennes de certification Les organismes de certification Les procédures de contrôles et de sécurités Les responsabilités civiles

12
Les Organismes notifiés
Les organismes notifiés sont des organismes désignés par les autorités administratives, compétentes pour le contrôle du respect des directives européennes. Ils interviennent lors des procédures d’évaluation de la conformité. La liste des organismes notifiés est publiée au Journal Officiel des Communautés Européennes. Il n’y a en France qu’un organisme notifié pour les dispositifs biomédicaux : le GIE-MED, regroupement des organismes LNE (Laboratoire National d’essai) et G-Med (Groupement pour l’Evaluation des Dispositifs Médicaux)
 et G-Med (Groupement pour l’Evaluation des Dispositifs Médicaux)")
13
Sommaire Procédures de certification du marquage CE
Directives et Normes européennes de certification Les organismes de certification Les procédures de contrôles et de sécurités Les responsabilités civiles

14
Procédures de sécurité
Dommage : cette notion est la même dans presque tout les domaines, dans le domaine médicale on la définie suivant le ISO/CEI Guide51 (1999) par : blessure physique ou une atteinte à la santé des personnes, ou dégât causé aux biens ou à l’environnement Risque : suivant la norme médicale ISO (2000) et ISO/CEI Guide 51 (1999) est défini par : Combinaison de la probabilité d’un dommage et de sa gravité C’est la probabilité qu’il y ait un décès ou des complications ou des effets secondaires. Sécurité : elle est définie comme la présentation de dégâts sur l’humain, le robot et les éléments avec lesquelles le robot interagie (Dhillon, 1991). Le terme sécurité employé dans le domaine médical pour exprimer un niveau de sécurité atteint en réduisant le risque à 1 niveau acceptable (ISO/CEI Guide51).
 par : blessure physique ou une atteinte à la santé des personnes, ou dégât causé aux biens ou à l’environnement. Risque : suivant la norme médicale ISO (2000) et ISO/CEI Guide 51 (1999) est défini par : Combinaison de la probabilité d’un dommage et de sa gravité. C’est la probabilité qu’il y ait un décès ou des complications ou des effets secondaires. Sécurité : elle est définie comme la présentation de dégâts sur l’humain, le robot et les éléments avec lesquelles le robot interagie (Dhillon, 1991). Le terme sécurité employé dans le domaine médical pour exprimer un niveau de sécurité atteint en réduisant le risque à 1 niveau acceptable (ISO/CEI Guide51).")
15
Gestion des risques

16
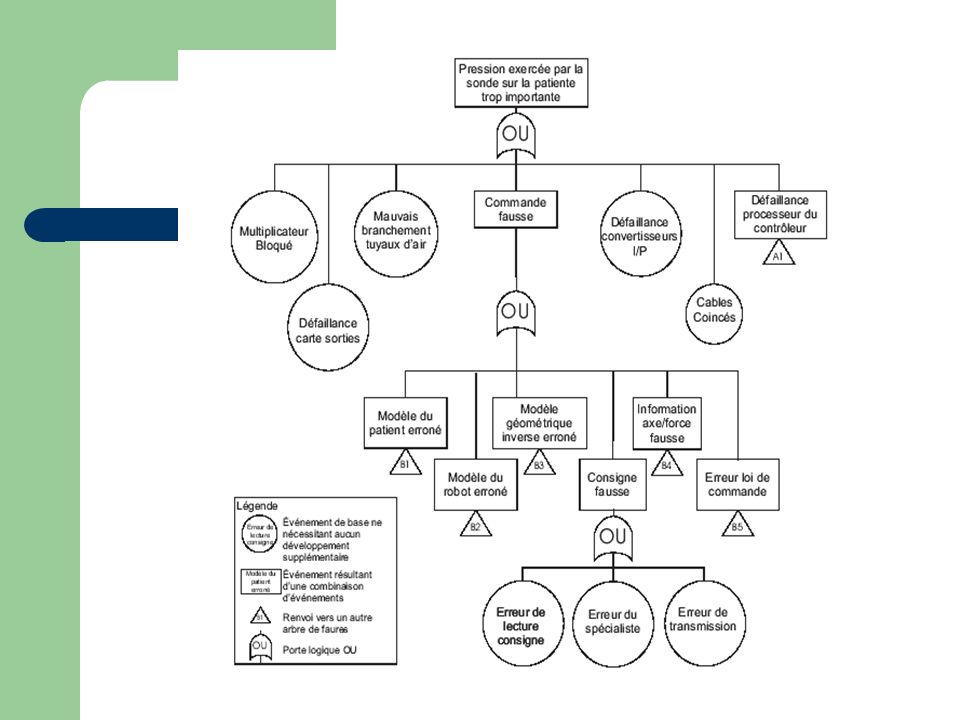
Techniques d’analyse du risque

18
Evaluation des risques

19
Evaluation des risques

20
Techniques de maîtrise de risque 1
La conception matérielle : on a la notion de sécurité intrinsèque lorsque la structure même du robot ne présente pas de phénomène dangereux (par exemple : pas de bord de tranchant) Sécurité autour des actionneurs qui se présente dans : la précision des mouvements articulaires, avoir un mécanisme d’arrêt d’urgence. sécurité de la redondance des composants : doubler le nombre des capteurs et des cartes de contrôle. Sécurité lors de la mise sous tension : spécifier l’étape de préparation du patient lors de mise en route du robot.
 Sécurité autour des actionneurs qui se présente dans : la précision des mouvements articulaires, avoir un mécanisme d’arrêt d’urgence. sécurité de la redondance des composants : doubler le nombre des capteurs et des cartes de contrôle. Sécurité lors de la mise sous tension : spécifier l’étape de préparation du patient lors de mise en route du robot.")
21
Techniques de maîtrise de risque 2
Surveillance du système : Les mécanismes de surveillance doivent en général (Leveson, 1995) : être indépendants des dispositifs qu’ils surveillent, détecter les problèmes de très bas niveau pouvant induire des situations dangereuses augmenter le moins possible la complexité du système, demander une maintenance simple. Conception des interfaces humain-machine : les interfaces fournissent au spécialiste des informations sur l’état du système mais surtout les données essentielles relatives à la tâche ( par exemple une image pour une opération chirurgicale ou pour une échographie).
 : être indépendants des dispositifs qu’ils surveillent, détecter les problèmes de très bas niveau pouvant induire des situations dangereuses. augmenter le moins possible la complexité du système, demander une maintenance simple. Conception des interfaces humain-machine : les interfaces fournissent au spécialiste des informations sur l’état du système mais surtout les données essentielles relatives à la tâche ( par exemple une image pour une opération chirurgicale ou pour une échographie).")
22
La matériovigilance 1 La surveillance des incidents ou des risques d'incidents peuvent résulter de l'utilisation des dispositifs médicaux après leur mise sur le marché. Procédure de matériovigilance : Signalement, enregistrement des incidents et des risques d’incidents Enregistrement, évaluation et exploitation des informations signalées dans un but de prévention. Réalisation de toutes les études et les travaux concernant la sécurité d’utilisation des robots. Réalisation et suivi des actions correctives décidées.

23
La matériovigilance 2 Obligation pour les établissements de santé de désigner un correspondant local de matériovigilance chargé de : l’enregistrement, l’analyse et la validation des signalements d’incidents mettant en cause un robot, et de recommander le cas échéant des mesures conservatoires. La transmission des déclarations d’incidents ou de risques d’incidents, sans délai pour les incidents graves, tout les trois mois pour des signalements facultatifs. L’aide à l’évaluation des données, le conseil aux déclarants et la sensibilisation des utilisateurs aux problèmes de matériovigilance. La conduite des travaux et enquêtes demandés par l’AFSSAPS

24
Sommaire Procédures de certification du marquage CE
Directives et Normes européennes de certification Les organismes de certification Les procédures de contrôles et de sécurités Les responsabilités civiles

25
Notion de fautes Généralement, on appelle faute, “l'acte que n'aurait pas commis un médecin normalement diligent et compétent”. Les fautes au sein d’un hôpital Les fautes au sein d’une clinique

26
Loi du 19/05/1998 Elle pose le principe de la responsabilité du constructeur ou du producteur, dès que sont constatés des défauts sur le produit ayant entraîné des dommages. La responsabilité du producteur est donc engagée en cas de problème. SAUF: si le produit na pas été mis en circulation dans le respect des normes (non marquage CE). Exemple: produit volé si le défaut était inexistant à la mise en circulation si l’état des connaissances scientifiques et techniques ne permettait pas de mettre le défaut en évidence (exemple: un robot fabriqué dans une matière composite dont on découvre plus tard la toxicité). si le produit n’est pas destiné à un usage commercial (exemple: utilisation d'un prototype) si la victime a elle-même commis une faute (exemple: patient blessé à la suite d’une erreur de manipulation sur un bras mécanique lui appartenant.)
. Exemple: produit volé si le défaut était inexistant à la mise en circulation si l’état des connaissances scientifiques et techniques ne permettait pas de mettre le défaut en évidence (exemple: un robot fabriqué dans une matière composite dont on découvre plus tard la toxicité). si le produit n’est pas destiné à un usage commercial (exemple: utilisation d un prototype) si la victime a elle-même commis une faute (exemple: patient blessé à la suite d’une erreur de manipulation sur un bras mécanique lui appartenant.)")
Présentations similaires