Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Apoptose Olivier Meilhac Unité INSERM 698 CHU X. Bichat Paris 18e

2
1. Introduction Apoptosis - (Gr. "falling") a process seen in multicellular organisms, by which specific cells are killed and removed for the benefit of the organism. Kerr, J.F.R., Wyllie, A.H. and Currie, A.R Br. J. Cancer 26:239. Apoptosis = chute des feuilles en automne
 a process seen in multicellular organisms, by which specific cells are killed and removed for the benefit of the organism. Kerr, J.F.R., Wyllie, A.H. and Currie, A.R Br. J. Cancer 26:239. Apoptosis = chute des feuilles en automne.")
3
Définition – rôle physiologique
« mort cellulaire programmée » homéostasie : évite l’excès de cellules éliminer les cellules potentiellement dangereuses celles qui gênent le développement événements inducteurs : La mort par apoptose peut être déclenchée dans plusieurs types de situation, dépendantes de l’environnement des cellules, des signaux qu’elles reçoivent ainsi que de leur état : a/ la cellule ne reçoit pas de signaux de survie (facteurs de croissance, contacts physiques avec son environnement : cellules ou matrice extracellulaire) b/ la cellule reçoit des signaux de mort : via un récepteur Fas ligand, TNF via une agression modérée (irradiations, poisons, LDLox, oxystérols, choc thermique…) c/ la cellule reçoit des signaux de prolifération incompatibles avec l’état général de la cellule (expression de certains gènes : p53, c-fos, c-myc, bcl- 2) signaux de prolifération incompatibles avec l’état cellulaire signaux de mort : perte des signaux de survie (facteurs de croissance, contacts) - récepteur-médiés - non récepteur médiés
 b/ la cellule reçoit des signaux de mort : via un récepteur Fas ligand, TNF. via une agression modérée (irradiations, poisons, LDLox, oxystérols, choc thermique…) c/ la cellule reçoit des signaux de prolifération incompatibles avec l’état général de la cellule (expression de certains gènes : p53, c-fos, c-myc, bcl- 2) signaux de prolifération incompatibles avec l’état cellulaire. signaux de mort : perte des signaux de survie (facteurs de croissance, contacts) - récepteur-médiés. - non récepteur médiés.")
4
Déséquilibre pathologie
prolifération apoptose prolifération apoptose SIDA Maladies dégénératives anémie aplasique troubles ischémiques (infarctus du myocarde…) maladies du foie induites par toxine (alcool…) Anémie aplasique : Forme d'anémie dans laquelle la moelle épinière ne produit pas un nombre suffisant d'éléments sanguins Cancer Maladies auto-immunes Infections virales
 maladies du foie induites par toxine (alcool…) Anémie aplasique : Forme d anémie dans laquelle la moelle épinière ne produit pas un nombre suffisant d éléments sanguins. Cancer. Maladies auto-immunes. Infections virales.")
5
Apoptose vs nécrose Nécrose: gonflement de la cellule
changements mitochondriaux destruction de la cellule inflammation ( rougeur, chaleur, douleur, tuméfaction) Relargage de cytokines ( => adhésion de cellules à l’endothélium, fièvre) => Recrutement de cellules blanches Apoptose : Changements dans le noyau, condensation Fragmentation du noyau Fragmentation de l’ADN ( visible si on l’extrait du noyau) Fragmentation de la cellule => corps apoptotiques Reconnaissance par des cellules phagocytaires qui éliminent les cellules en apoptose. Si ce n’est pas le cas => nécrose apoptotique
 Relargage de cytokines ( => adhésion de cellules à l’endothélium, fièvre) => Recrutement de cellules blanches. Apoptose : Changements dans le noyau, condensation. Fragmentation du noyau. Fragmentation de l’ADN ( visible si on l’extrait du noyau) Fragmentation de la cellule => corps apoptotiques. Reconnaissance par des cellules phagocytaires qui éliminent les cellules en apoptose. Si ce n’est pas le cas => nécrose apoptotique.")
6
Death Receptor Pathway Mitochondrial Pathway
FasL oxidants ceramide others Fas/Apo1 /CD95 DNA damage D D Bcl-2 D D D FADD DISC Procaspase 8 dATP Apaf -1 BID Procaspase 9 Cytochrome c Caspase 8 Procaspase 3 dATP 2 voies : Récepteur de la mort et voie mitochondriale pour déclencher l’apoptose. Ces deux voies sont connectées. Elles mènent toutes les deux à l’activation de protéases notamment la caspase 3. Apaf -1 Caspase 9 Cellular targets Caspase 3 apoptosome Hengartner, M.O Nature. 407:770. Green, D. and Kroemer, G Trends Cell Biol. 8:267.

7
2. Voie extrinsèque Les récepteurs de mort
TNF-R1 TNF Fas FasL DR3 Apo3L DR4 Apo2L/TRAIL DR5 Apo2L/TRAIL CAR 1 ? Elimination des cellules en excès ou dangereuses : développement apprentissage du système immunitaire cellules tumorales DD DD DD

8
TNF FasL TNFR1 Fas DISC TRADD FADD Pro-caspase 8 caspase 8 active
DED Pro-caspase 8 Récepteurs de la mort: Voie extrinsèque activée par un signal extracellulaire: liaison Ligand-Récepteur Ex: TNF-R1/TNF alpha => trimérisation du R, rapprochement de domaines intracellulaires, recrutement de protéines adaptatrices (TRADD), d’un complexe DISC, qui recrute lui-même des caspases ( initialement pro-caspase 8) Ex FasL/ Fas: si pro-caspase 8 fixée au DISC, elle s’active en caspase 8. Caspases = protéases qui clivent d’autres protéases et ainsi de suite, jusqu’aux caspases effectrices qui vont cliver des substrats à l’intérieur de la cellule. caspase 8 active
, d’un complexe DISC, qui recrute lui-même des caspases ( initialement pro-caspase 8) Ex FasL/ Fas: si pro-caspase 8 fixée au DISC, elle s’active en caspase 8. Caspases = protéases qui clivent d’autres protéases et ainsi de suite, jusqu’aux caspases effectrices qui vont cliver des substrats à l’intérieur de la cellule. caspase 8 active.")
9
Les cellules tumorales contre-attaquent
LT augmente l’expression de Fas L, l’expose à sa surface pour tenter d’éliminer la cellule tumorale. Toutes les cellules de l’organisme expriment Fas, y compris les cellules tumorales, mais elles sont résistantes. Par ailleurs elles vont se mettre à exprimer le FasL, qui va induire la mort par apoptose du LT. => Arme de défense des cellules tumorales 2004, 4:321-6 Le lymphocyte T reconnaît un antigène tumoral (CMH) Il augmente l’expression de FasL, mais la cellule tumorale est résistante Lymphocyte T activé exprimant Fas, c’est lui qui meurt FasL métastase : survie des cellules en milieu immunologique hostile ?
 Il augmente l’expression de FasL, mais la cellule tumorale est résistante. Lymphocyte T activé exprimant Fas, c’est lui qui meurt. FasL métastase : survie des cellules en milieu immunologique hostile")
10
3.Voie intrinsèque Formation de l’apoptosome
Elimination des cellules en réponse à une agression modérée : radiations ionisantes chimiothérapie dommages mitochondriaux : oxydants, calcium, céramide Formation de l’apoptosome Fig. 2. Model for activation of Apaf-1 and procaspase-9. (a) Successive binding of cytochrome c (cyt c) and dATP/ATP converts Apaf-1 from a ‘closed’ monomer to an ‘open’ conformer and then to a heptameric platform for procaspase-9 assembly [41]. (b) An inactive procaspase-9 monomer on one spoke of the apoptosome is presumed to recruit another monomer to create the asymmetric dimer having a single active site (see text for full details). Voie intrinsèque : La mitochondrie reçoit des signaux de l’environnement ( ex: radiations, poisons, stress oxydatifs …) Elle envoie ensuite des signaux de morts: libération de cytochrome C, il s’associe à Apaf-1, Apaf-1 se déplie et libère des domaines pour former des heptamères de Apaf-1,hepatmères qui recrutent de la pro-caspase 9, qui s’active=> cette cascade rejoint la caspase 3 aussi Dec (6):715-20
 Successive binding of cytochrome c (cyt c) and dATP/ATP converts Apaf-1 from a ‘closed’ monomer to an ‘open’ conformer and then to a heptameric platform for procaspase-9 assembly [41]. (b) An inactive procaspase-9 monomer on one spoke of the apoptosome is presumed to recruit another monomer to create the asymmetric dimer having a single active site (see text for full details). Voie intrinsèque : La mitochondrie reçoit des signaux de l’environnement ( ex: radiations, poisons, stress oxydatifs …) Elle envoie ensuite des signaux de morts: libération de cytochrome C, il s’associe à Apaf-1, Apaf-1 se déplie et libère des domaines pour former des heptamères de Apaf-1,hepatmères qui recrutent de la pro-caspase 9, qui s’active=> cette cascade rejoint la caspase 3 aussi. Dec (6):")
11
Relargage de DIABLO, qui se complexe avec IAP ( complexe inhibant l’apoptose) il est alors inactif=> neutralisation des protéines anti-apototiques
 il est alors inactif=> neutralisation des protéines anti-apototiques")
12
activent floppase inhibent translocase
-Relargage de facteurs pro-apototiques, ex: AIF ( facteur induisant l’apoptose) active les floppases et inhibe les translocases( flip-flop) Libération d’endonucléases qui fragmentent l’ADN Externalisation des phospholipides ( phosphatidyle sérine) qui permet la reconnaissance par les macrophages
 active les floppases et inhibe les translocases( flip-flop) Libération d’endonucléases qui fragmentent l’ADN. Externalisation des phospholipides ( phosphatidyle sérine) qui permet la reconnaissance par les macrophages.")
13
Voie extrinsèque voie intrisèque
Figure 1. Apoptosis signalling via the Fas receptor. Binding of FasL to Fas induces the recruitment of FADD and pro-caspase-8 to the cytoplasmic tail of Fas, and the formation of the DISC. At the DISC, caspase-8 is activated. In type I cells, sufficient caspase-8 is generated to activate pro-caspase-3 directly. However, in type II cells, activation of pro-caspase-3 occurs indirectly through cleavage and activation of Bid. Truncated Bid (tBid) triggers the release of pro-apoptotic molecules from the intermembrane space of mitochondria. Released cytochrome c (cyto c) clusters with Apaf-1 and pro-caspase-9 in the presence of dATP to activate caspase-9. Activated caspase-9 cleaves and activates caspase-3, triggering a caspase cascade, which ultimately results in the death of the cell. Les 2 voies ne sont pas comlètement indépendantes. Liaison FasL-Fas pas toujours suffisante pour activer assez de caspases 8. Il faut que simultanément il y ait un signal mitochondrial. Bid peut être clivé par une caspase 8, qui se lie ensuite à Bax. Caspase 9 activée par l’apotosome est capable aussi d’activer la caspase . => Caspase 3, protéine détectée pour savoir si une cellule est en apoptose
 triggers the release of pro-apoptotic molecules from the intermembrane space of mitochondria. Released cytochrome c (cyto c) clusters with Apaf-1 and pro-caspase-9 in the presence of dATP to activate caspase-9. Activated caspase-9 cleaves and activates caspase-3, triggering a caspase cascade, which ultimately results in the death of the cell. Les 2 voies ne sont pas comlètement indépendantes. Liaison FasL-Fas pas toujours suffisante pour activer assez de caspases 8. Il faut que simultanément il y ait un signal mitochondrial. Bid peut être clivé par une caspase 8, qui se lie ensuite à Bax. Caspase 9 activée par l’apotosome est capable aussi d’activer la caspase . => Caspase 3, protéine détectée pour savoir si une cellule est en apoptose.")
14
4. Famille Bcl-2 BH : Bcl-2 Homology BH4 : anti-apoptotique
Curr Opin Cell Biol Dec;15(6):691-9. BH : Bcl-2 Homology BH4 : anti-apoptotique BH1-3 : pro-apoptotiques BH3-only: pro-apoptotiques indirects Activent les pro-apoptotiques Inhibent les anti-apoptotiques Ex : Bad –Bik : inactivent Bcl-2 et BclXL Bid- Bim : activent Bax et Bak Bcl2 : B cell lymphom ( découvert dans lynphome avec translocation ) Famille très nombreuse Bcl 2 ( la protéine) : anti apoptotique Protéines avec domaine BH4 => anti-apoptotiques Protéines avec domaine BH1-3 => pro-apoptotiques Ex: Bax peut se complexer avec Bcl2 et inhiber son activité anti-apoptotique
: BH : Bcl-2 Homology. BH4 : anti-apoptotique. BH1-3 : pro-apoptotiques. BH3-only: pro-apoptotiques indirects. Activent les pro-apoptotiques. Inhibent les anti-apoptotiques. Ex : Bad –Bik : inactivent Bcl-2 et BclXL. Bid- Bim : activent Bax et Bak. Bcl2 : B cell lymphom ( découvert dans lynphome avec translocation ) Famille très nombreuse. Bcl 2 ( la protéine) : anti apoptotique. Protéines avec domaine BH4 => anti-apoptotiques. Protéines avec domaine BH1-3 => pro-apoptotiques. Ex: Bax peut se complexer avec Bcl2 et inhiber son activité anti-apoptotique.")
15
BID Truncated BID Caspase 8
Bcl 2 : bouche les pores de la mitochondrie pour maintenir le cytochrome c à l’intérieur. Peut s’associer à Bax, dans ce cas Bcl 2 est dans le cytoplasme, il ne bouche plus le pore => libération du cytochrome c

16
p53 Arrêt du cycle cellulaire
p53 : pro-apoptotique ou arrêt du cycle cellulaire Déficit en facteurs de croissance c-myc Lésions de l’ADN p53 transcription + Bax + p21waf + Fas, DR5 + cycline G + PUMA/NOXA + gadd45 + APAF1 + mdm2 - Bcl-2… P53: favorise la transcription de gènes pro-apoptotiques: ex Bax, APAF-1. Bcl2 va être inhibé. Arrête le cycle cellulaire. apoptose Arrêt du cycle cellulaire

17
Si dimères Bcl2-Bcl2 => bouchent les pores des mitochondries
Si trop de Bcl2 => prolifération et accumulation de cellules Si trop de Bax => trop d’apoptose Si accumulation de cellules mutantes par perte de controle apoptotique (ex: bcl-2 augmentée, mutation p53) formation de tumeur
 formation de tumeur.")
18
5. Caspases effectrices ou exécutrices
Pas de prodomaine Nter (dépourvu d’activité enzymatique) 3, 6, 7 responsables des événements destructeurs caspase cascade Signaux permettent recrutement de pro-caspases ( ex: 8, 9), elles s’activent, elles reconnaissent ensuite les caspases effectrices : 3, 6, 7. Avant l’activation des caspases effectrices, pas de dommages cellulaires, le processus est encore réversible.
 3, 6, 7. responsables des événements destructeurs. caspase cascade. Signaux permettent recrutement de pro-caspases ( ex: 8, 9), elles s’activent, elles reconnaissent ensuite les caspases effectrices : 3, 6, 7. Avant l’activation des caspases effectrices, pas de dommages cellulaires, le processus est encore réversible.")
19
Substrats des caspases
65 en 1998 : plus de 280 en 2003 protéines de structure (cytoplasme et noyau) transduction de signal transcription / régulation contrôle du cycle cellulaire réplication / réparation de l’ADN spécifiques de certains types cellulaires conséquence du clivage souvent inconnu clivage parfois tardif, (accessoire, conséquence de la cascade d’activation) Substrats des caspases: Protéines de structure => cellule perd sa forme, déformation Cell Death and Differentiation, 2003; 10:76-100
 transduction de signal. transcription / régulation. contrôle du cycle cellulaire. réplication / réparation de l’ADN. spécifiques de certains types cellulaires. conséquence du clivage souvent inconnu. clivage parfois tardif, (accessoire, conséquence de la cascade d’activation) Substrats des caspases: Protéines de structure => cellule perd sa forme, déformation. Cell Death and Differentiation, 2003; 10:")
20
Substrats en rapport avec les changements morphologiques
ICAD : lève l’inhibition d’une DNAse fragmentation de l’ADN ADN hélicase condensation de la chromatine, remodelage nucléaire gelsoline dépolymérisation de l’actine F kinases ROCK-1, PAK2 P° chaine légère de la myosine filaments intermédiaires : cytokératine 18, vimentine organisation des filaments : gas-2, plectine « tenségrité » : liaison cellules-cellules, cellules -matrice extracellulaire Fodrine, FAK, paxilline inh° signaux de survie via intégrines b-caténine, E-cadhérine, plakoglobine, desmogléine… Condensation cellulaire, rétraction, détachement protéines du RE, Golgi, vésicules de transport protéines nucléaires lamina, pores (transport perturbé) réparation de l’ADN : PARP-1… protéines stabilisant la chromatine RNA hélicase A, facteurs de splicing no transcription Facteurs d’initiation de traduction eIF2,3,4… « blebbing » Bien retenir ce qui est en rouge ! Perturbation du transport des protéines, de la transcrition et de la traduction Modifications dans la structure même de la cellule -> fragmentation de l’ADN, Dépolymérisation de l’actine F -> perte de structure de la membrane, invaginations
 réparation de l’ADN : PARP-1… protéines stabilisant la chromatine. RNA hélicase A, facteurs de splicing no transcription. Facteurs d’initiation de traduction eIF2,3,4… « blebbing » Bien retenir ce qui est en rouge ! Perturbation du transport des protéines, de la transcrition et de la traduction Modifications dans la structure même de la cellule -> fragmentation de l’ADN, Dépolymérisation de l’actine F -> perte de structure de la membrane, invaginations.")
21
Substrats des caspases et signalisation
protéines antiapoptotiques : kinases : Bcl-2, BclX-L : antiproapoptotiques c-FLIP : inhibiteur de la caspase 8 Bid c-Bid : libération de cytochrome c virus : leurres Bcl-2 like Akt et Raf-1 phosphorylent et inactivent Bad NFK-B : fragment de p65 lie l’ADN mais pas de transactivation IK-B : fragment n’est plus dégradé par le protéasome MEKK1 :fragment active d’autres caspases Des caspases « grignotent » le domaine BH4 de Bcl2, le rendant ainsi pro-apoptotique. Confortent la cellule dans la voie apoptotique.

22
6. Méthodes de détection de l’apoptose

24
Changements morphologiques
control apoptotic Microscopie photonique : - condensation de l’ADN, fragmentation nucléaire, corps apoptotiques Microscopie électronique : - balayage - transmission Microscope photonique: Condensation de l’ADN => + de coloration du noyau ME à balayage : 2 images en haut à droite: on voit les invaginations de la membranes ( « blebbing ») ME à transmission: permet de voir ce qui se passe à l’intérieur de la cellule: margination de la chromatine avec condensation.
 ME à transmission: permet de voir ce qui se passe à l’intérieur de la cellule: margination de la chromatine avec condensation.")
25
Fragmentation de l’ADN
Agarose gel electrophoresis Fragmentation inter-nucléosomale de l’ADN : très caractéristique de l’apoptose. Endonucléases coupent entre les nucléosomes => longueurs d’ADN vont être des multiples de 180 à 240 paires de base => quand on fait migrer de l’ADN, on va obtenir des « ladder » ( des bandes en échelle). Pour faire migrer l’ADN, on utilise un gel d’agarose, qui polymérise, il y a des pores dans ce gel. On applique une différence de potentiel entre le haut et le abs du gel. L’ADN étant chargé négativement, on met l’anode (pôle +) en bas, pour l’attirer vers le bas. Pour visualiser l’ADN, on utilise du bromure d’éthidium. A droite : « smear » traînée d’ADN de la nécrose. A gauche : « ladder » d’ADN de l’apoptose Chromatin Fragmented Chromatin
. Pour faire migrer l’ADN, on utilise un gel d’agarose, qui polymérise, il y a des pores dans ce gel. On applique une différence de potentiel entre le haut et le abs du gel. L’ADN étant chargé négativement, on met l’anode (pôle +) en bas, pour l’attirer vers le bas. Pour visualiser l’ADN, on utilise du bromure d’éthidium. A droite : « smear » traînée d’ADN de la nécrose. A gauche : « ladder » d’ADN de l’apoptose. Chromatin Fragmented. Chromatin.")
26
Méthode TUNEL: Terminal transferase dUTP Nick End Labelling
La terminal transférase permet incorporer des dUTP marqués à chaque fois qu’il y a un bord franc Marque les noyaux en apoptose. Noyaux en apoptose marqués en brun sur image en haut à droite. Souvent on utilise un Ac primaire, qui reconnaît l’Ag qui nous intéresse. Ensuite on a un Ac secondaire couplé (à la peroxydase par exemple; qui oxyde son substrat et le rend marron).
.")
27
Activation des caspases, changements mitochondriaux
substrats chromogènes spécifiques anticorps spécifiques : western blot, immunodétection in situ western-blot cytochrome c : extraits cytosoliques / mitochondriaux cytométrie On peut aussi évaluer l’activation des caspases et les changements mitochondriaux Il existe des substrats de caspases ( séquence en AA) qui une fois clivés, génèrent de la couleur. Chromogène On broie une cellule, on fait un extrait protéique pour le substrat rencontre la caspase. Western-blot: Electrophorèse: migration dans un gel de protéines en fonction du poids moléculaire. Les + grosses protéines ont du mal à se mouvoir. => permet de séparer les proétines Les protéines peuvent être chargées + ou -, pour annuler ce paramètre, on les charge toutes artificiellement - avec du SDS. Le SDS déplie aussi les protéines, elles sont alors linéaires. Une fois que les protéines ont migré sur le gel, on les transfère sur une membrane pour qu’elles soient accessibles à des Ac. « blot » = transfert sur membrane « western » en référence à Southern qui a découvert cette technique de transfert du gel à la membrane. On attire les protéines sur la membrane avec un champ +. On utilise ensuite un Ac spécifique qui reconnaît la protéine. Ex: pro-caspase 3 ( 17kD) et caspase 3 ( 23kD). L’Ac va reconnaître à la fois la pro-caspase 3 et la caspase 3, donc pour savoir si la cellule est en apoptose ou pas on a besoin de connaître son poids moléculaire. Immunodétection in situ: Détection de la caspase active. On utilise un Ac spécifique qui reconnaît les caspases actives, il reconnaît les zones de clivage. Western blot cytochrome C: il faut séparer le cytosol des organelles, on arrive à fractionner les compartiments cellulaires. Cytométrie: cellulent passent une à une devant un laser ou une lumière normale. Donne des infos sur la taille et la granulométrie.
 qui une fois clivés, génèrent de la couleur. Chromogène. On broie une cellule, on fait un extrait protéique pour le substrat rencontre la caspase. Western-blot: Electrophorèse: migration dans un gel de protéines en fonction du poids moléculaire. Les + grosses protéines ont du mal à se mouvoir. => permet de séparer les proétines. Les protéines peuvent être chargées + ou -, pour annuler ce paramètre, on les charge toutes artificiellement - avec du SDS. Le SDS déplie aussi les protéines, elles sont alors linéaires. Une fois que les protéines ont migré sur le gel, on les transfère sur une membrane pour qu’elles soient accessibles à des Ac. « blot » = transfert sur membrane. « western » en référence à Southern qui a découvert cette technique de transfert du gel à la membrane. On attire les protéines sur la membrane avec un champ +. On utilise ensuite un Ac spécifique qui reconnaît la protéine. Ex: pro-caspase 3 ( 17kD) et caspase 3 ( 23kD). L’Ac va reconnaître à la fois la pro-caspase 3 et la caspase 3, donc pour savoir si la cellule est en apoptose ou pas on a besoin de connaître son poids moléculaire. Immunodétection in situ: Détection de la caspase active. On utilise un Ac spécifique qui reconnaît les caspases actives, il reconnaît les zones de clivage. Western blot cytochrome C: il faut séparer le cytosol des organelles, on arrive à fractionner les compartiments cellulaires. Cytométrie: cellulent passent une à une devant un laser ou une lumière normale. Donne des infos sur la taille et la granulométrie.")
28
Exposition des phosphatidyl-sérines
A- control B-spontaneous apoptosis C- actinomycin D D- calcium ionophore Quandrants 1 – viable cells 2 – early apoptosis; PS exposed, intact membrane 3 – late apoptosis; PS exposed, disrupted membrane 4 – necrosis; disrupted & membrane loss 4 3 1 2 Propidium iodide Actinomycine D : inhibe la transcription et donc favorise l’apoptose Cytométrie en flux Abscisse: quantité de fluorescence complexée à l’Annexin V Ordonnée: quantité de fluorescence complexée à l’iodure de propidium. Annexine V, se fixe à la phosphatidylsérine si exposée à la membrane. Propidium rentre dans la cellule seulement s’il y a des trous dans la membrane = cellules en nécrose A: cellules vivantes peu d’annexineV et pas de propidium B: bcp d’annexine V, déplacement à droite. => apoptose principalement D: 4/ cellules en nécrose post apoptotique Annexin V-FITC

29
Abréviations : TNF: Tumor Necrosis Factor TRADD: TNF Receptor-Associated Death Domain TRAF2 : TNF Receptor-Associated Factor 2 RIP: Receptor-Interacting Protein SODD: Silencer Of Death Domains FADD: Fas-Associated Death Domain DISC: Death Inducing Signaling Complex TRAIL: TNF Related Apoptosis Inducing Ligand DR: Death Receptor IAP : Inhibitor of Apoptosis Protein FLICE : FADD Like Interleukin-1b Converting Enzyme (caspase 8) cFLIP: FLICE Inhibitory Protein APAF: Apoptosis Protease Activating Factor BH: Bcl-2 Homology Bcl-2: B Cell Lymphoma 2
 cFLIP: FLICE Inhibitory Protein. APAF: Apoptosis Protease Activating Factor. BH: Bcl-2 Homology. Bcl-2: B Cell Lymphoma 2.")
30
7. Anoïkis : apoptose par détachement
Quand la cellules perd ses contacts par les intégrines, elle perd des signaux de survie.

32
Signaux de survie a b Akt Collagène, fibronectine, vitronectine
Cellule 2 Collagène, fibronectine, vitronectine Facteurs de croissance INTEGRINES RTK (ex: TrkB) Cadhérines membrane plasmique a b Cellule 1 Protéines liant l’actine Pinch, Nck2, IRS-1 ? actine ILK FAK Protéines adaptatrices b-caténine PI3K Récepteurs qui ressentent la matrice extra cellulaire, la présence de vitronectine, collagène, facteurs de croissance… Intégrines => voies de survie: PI3K, Akt Si perte des intégrines => perte de ces signaux anti-apoptotiques Contacts cellule-cellules: cadhérines Cytosquelette Akt Signaux anti-apoptotiques : Inhibition de Bad, caspase 9 Transcription d’IAP, bcl-2… Inhibition de la transcription de FasL, bim…
 Cadhérines. membrane. plasmique. a. b. Cellule 1. Protéines liant l’actine. Pinch, Nck2, IRS-1 actine. ILK. FAK. Protéines adaptatrices. b-caténine. PI3K. Récepteurs qui ressentent la matrice extra cellulaire, la présence de vitronectine, collagène, facteurs de croissance… Intégrines => voies de survie: PI3K, Akt. Si perte des intégrines => perte de ces signaux anti-apoptotiques. Contacts cellule-cellules: cadhérines. Cytosquelette. Akt. Signaux anti-apoptotiques : Inhibition de Bad, caspase 9. Transcription d’IAP, bcl-2… Inhibition de la transcription de FasL, bim…")
33
gènes pro-apoptotiques gènes anti-apoptotiques
AKT - + - Caspase 9 Bad FoxO YAP JNK/p38 iKB-kinase CREB mdm2 P P P 14.3.3 14.3.3 14.3.3 Bcl-2 E3 Ubiquitine-ligase Dégradation d’IKB séquestrés dans le cytosol Dégradation de p53 Translocation de NFKB NFKB FoxO Transcription de gènes pro-apoptotiques YAP p73 FasL, bim… Transcription de gènes anti-apoptotiques bcl-2, IAP… NOYAU

34
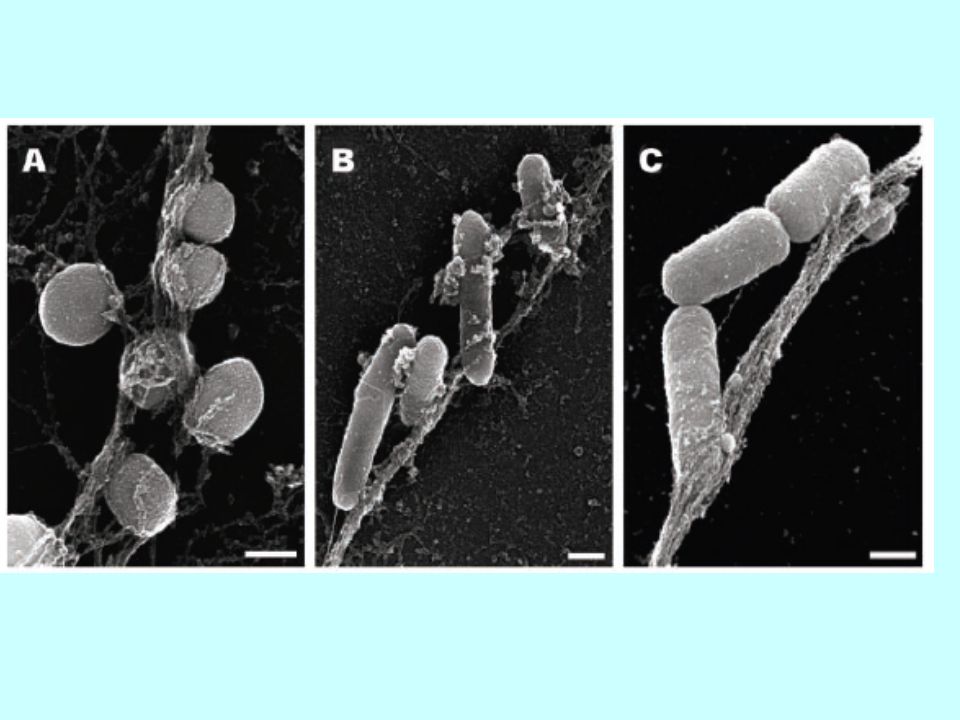
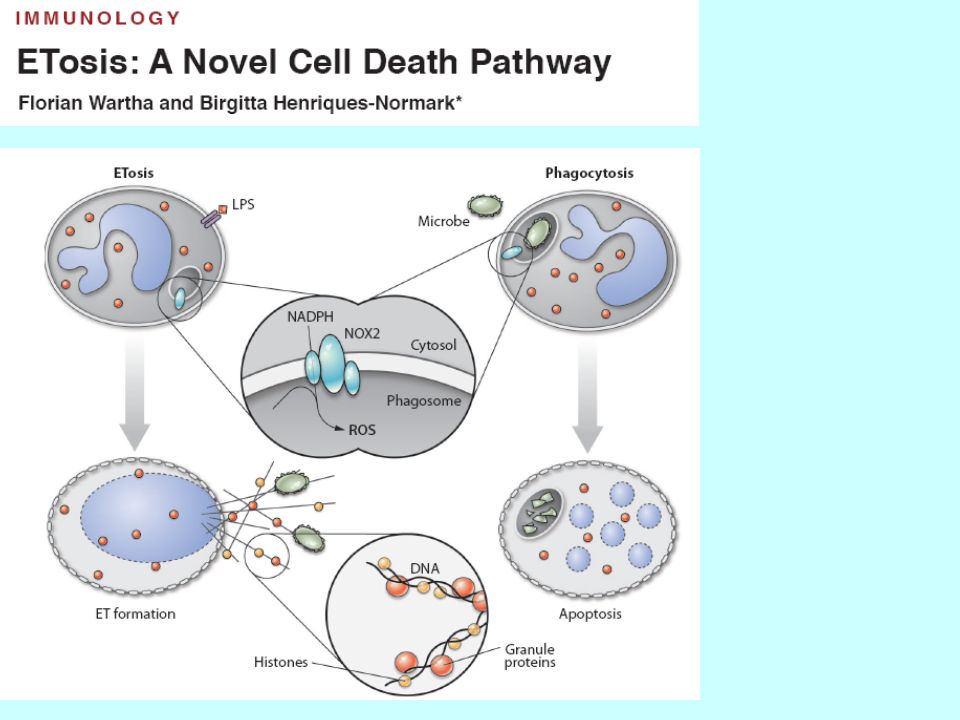
8. Nouvelles formes d’apoptose
8.1 ETosis Etosis Neutrophiles expulsent leur ADN => filets d’ADN qui attrapent des bactérie. L’ADN s’associe aux enzymes des neutrophiles qui sont bactéricides et pro-coagulantes.

35
Elastase DAPI Histones Elastase Histones

38
8.1 Eryptosis Erythrocyte Programmed Cell Death (Eryptosis)
Introduction: Suicidal death of erythrocytes (eryptosis) is characterized by cell shrinkage, membrane blebbing, activation of proteases and phosphatidylserine exposure at the outer membrane leaflet. Phosphatidylserine at the erythrocyte surface is recognised by macrophages which engulf and degrade the affected cells. Eryptosis is triggered by erythrocyte injury following several stressors including osmotic shock, oxidative stress, ligation of cell membrane antigens and energy depletion. Two signalling pathways have been identified so far which converge to trigger eryptosis. On the one hand, the formation of prostaglandin E2 through activation of cyclooxygenase leads to activation of a Ca2+-permeable cation channel. On the other hand, the activation of a phospholipase A2 leads to release of platelet activating factor (PAF), which in turn activates a sphingomyelinase leading to formation of the proapoptotic sphingolipid ceramide. The increased cytosolic Ca2+ concentrations and the elevated ceramide levels lead to scrambling of the cell membrane with subsequent shift of phosphatidylserine from the inner to the outer leaflet of the erythrocyte membrane. Moreover, Ca2+ activates Ca2+-sensitive K+ channels leading to cellular KCl loss and cell shrinkage. In addition, Ca2+ stimulates the protease calpain resulting in degradation of the cytoskeleton. Eryptosis is inhibited by erythropoietin which thus extends the life span of circulating erythrocytes. Eryptosis may be a mechanism of defective erythrocytes to escape hemolysis. On the other hand, excessive eryptosis favours the development of anemia. Conditions with excessive eryptosis include iron deficiency, lead or mercury intoxication, sickle cell anemia, thalassemia, and glucose-6- phosphate dehydrogenase deficiency, malaria and infection with hemolysin-forming pathogens. Cyclooxygenase activation, PGE2 formation and activation of cation channels: Hyperosmotic shock and Cl--removal trigger the release of prostaglandin E2 (PGE2). PGE2, in turn, activates nonselective cation channels, increases the cytosolic Ca2+ concentration, and stimulates phosphatidylserine exposure at the erythrocyte surface. Accordingly, osmotic cell shrinkage activates the cation channels and triggers erythrocyte Ca2+ uptake. The same or similar channels are activated by oxidative stress which similarly triggers Ca2+ entry. The channels are inhibited by intracellular or extracellular Cl-, and the activation of the cation channels by Cl--removal is abolished by the cyclooxygenase inhibitor diclophenac. Moreover, the phospholipase-A2 inhibitors quinacrine and palmitoyl-trifluoromethylketone and the cyclooxygenase inhibitors acetylsalicylic acid and diclophenac blunt the increase of phosphatidylserine exposure following Cl- removal. PGE2 further activates the Ca2+-dependent cysteine endopeptidase calpain, an effect, however, apparently not required for stimulation of phosphatidylserine exposure but playing a role in the degradation of the cytoskeleton. Moreover, increased Ca2+ leads to membrane vesiculation and subsequently to a decrease of the erythrocyte volume. Energy depletion impairs the replenishment of GSH and thus weakens the antioxidative defence of the erythrocytes. Accordingly, energy depletion similarly activates the cation channels. The cation channel inhibitors amiloride and ethylisopropylamiloride (EIPA) blunt the phosphatidylserine exposure following osmotic shock. Thus, activation of the cell volume- and oxidant-sensitive cation channel and subsequent Ca2+ entry contribute to the stimulation of erythrocyte scramblase. Besides its effect on phosphatidylserine scrambling an increase of cytosolic Ca2+ stimulates the Ca2+-sensitive "Gardos" K+ channels in erythrocytes. The subsequent hyperpolarization of the cell membrane drives Cl- in parallel to K+ out of the cell. The cellular loss of KCl favours cell shrinkage and contributes to the triggering of eryptosis. PAF formation and stimulation of sphingomyelinase: Beyond its effect on PGE2 formation, erythrocyte shrinkage triggers the formation of platelet activating factor (PAF), which is involved in the regulation of inflammation, thrombosis, atherogenesis and cardiovascular function. PAF then stimulates a sphingomyelinase leading to the breakdown of sphingomyelin and release of ceramide from erythrocytes. Osmotic shock thus leads to the appearance of ceramide at the erythrocyte surface. At least partially due to ceramide formation, PAF triggers scrambling of the cell membrane with phosphatidylserine exposure at the erythrocyte surface. C6-ceramide as well as treatment with purified, bacterial sphingomyelinase similarly triggers phosphatidylserine scrambling. Moreover, eryptosis following osmotic shock is blunted by the sphingomyelinase inhibitor 3,4-dichloroiso-coumarin. The stimulation of phosphatidylserine exposure is also blunted by genetic knockout of PAF receptors (PAF receptor -/- mice), and by the PAF receptor antagonist ABT491. PAF further activates Ca2+-sensitive K+ channels ("Gardos" channels) in the erythrocyte cell membrane by sensitising them for the stimulating effects of cytosolic Ca2+. The signaling through PAF does, however, not necessarily require elevated cytosolic Ca2+ concentrations, and enhanced PAF levels at least partially account for Ca2+-independent, ceramide-mediated eryptosis. Chez les oiseaux il y a un noyau dans les globules rouges. Eryptosis: externalisation des phosphatidylsérine Augmentation du calcium et du stress oxydant.
 is characterized by cell shrinkage, membrane blebbing, activation of proteases and phosphatidylserine exposure at the outer membrane leaflet. Phosphatidylserine at the erythrocyte surface is recognised by macrophages which engulf and degrade the affected cells. Eryptosis is triggered by erythrocyte injury following several stressors including osmotic shock, oxidative stress, ligation of cell membrane antigens and energy depletion. Two signalling pathways have been identified so far which converge to trigger eryptosis. On the one hand, the formation of prostaglandin E2 through activation of cyclooxygenase leads to activation of a Ca2+-permeable cation channel. On the other hand, the activation of a phospholipase A2 leads to release of platelet activating factor (PAF), which in turn activates a sphingomyelinase leading to formation of the proapoptotic sphingolipid ceramide. The increased cytosolic Ca2+ concentrations and the elevated ceramide levels lead to scrambling of the cell membrane with subsequent shift of phosphatidylserine from the inner to the outer leaflet of the erythrocyte membrane. Moreover, Ca2+ activates Ca2+-sensitive K+ channels leading to cellular KCl loss and cell shrinkage. In addition, Ca2+ stimulates the protease calpain resulting in degradation of the cytoskeleton. Eryptosis is inhibited by erythropoietin which thus extends the life span of circulating erythrocytes. Eryptosis may be a mechanism of defective erythrocytes to escape hemolysis. On the other hand, excessive eryptosis favours the development of anemia. Conditions with excessive eryptosis include iron deficiency, lead or mercury intoxication, sickle cell anemia, thalassemia, and glucose-6- phosphate dehydrogenase deficiency, malaria and infection with hemolysin-forming pathogens. Cyclooxygenase activation, PGE2 formation and activation of cation channels: Hyperosmotic shock and Cl--removal trigger the release of prostaglandin E2 (PGE2). PGE2, in turn, activates nonselective cation channels, increases the cytosolic Ca2+ concentration, and stimulates phosphatidylserine exposure at the erythrocyte surface. Accordingly, osmotic cell shrinkage activates the cation channels and triggers erythrocyte Ca2+ uptake. The same or similar channels are activated by oxidative stress which similarly triggers Ca2+ entry. The channels are inhibited by intracellular or extracellular Cl-, and the activation of the cation channels by Cl--removal is abolished by the cyclooxygenase inhibitor diclophenac. Moreover, the phospholipase-A2 inhibitors quinacrine and palmitoyl-trifluoromethylketone and the cyclooxygenase inhibitors acetylsalicylic acid and diclophenac blunt the increase of phosphatidylserine exposure following Cl- removal. PGE2 further activates the Ca2+-dependent cysteine endopeptidase calpain, an effect, however, apparently not required for stimulation of phosphatidylserine exposure but playing a role in the degradation of the cytoskeleton. Moreover, increased Ca2+ leads to membrane vesiculation and subsequently to a decrease of the erythrocyte volume. Energy depletion impairs the replenishment of GSH and thus weakens the antioxidative defence of the erythrocytes. Accordingly, energy depletion similarly activates the cation channels. The cation channel inhibitors amiloride and ethylisopropylamiloride (EIPA) blunt the phosphatidylserine exposure following osmotic shock. Thus, activation of the cell volume- and oxidant-sensitive cation channel and subsequent Ca2+ entry contribute to the stimulation of erythrocyte scramblase. Besides its effect on phosphatidylserine scrambling an increase of cytosolic Ca2+ stimulates the Ca2+-sensitive Gardos K+ channels in erythrocytes. The subsequent hyperpolarization of the cell membrane drives Cl- in parallel to K+ out of the cell. The cellular loss of KCl favours cell shrinkage and contributes to the triggering of eryptosis. PAF formation and stimulation of sphingomyelinase: Beyond its effect on PGE2 formation, erythrocyte shrinkage triggers the formation of platelet activating factor (PAF), which is involved in the regulation of inflammation, thrombosis, atherogenesis and cardiovascular function. PAF then stimulates a sphingomyelinase leading to the breakdown of sphingomyelin and release of ceramide from erythrocytes. Osmotic shock thus leads to the appearance of ceramide at the erythrocyte surface. At least partially due to ceramide formation, PAF triggers scrambling of the cell membrane with phosphatidylserine exposure at the erythrocyte surface. C6-ceramide as well as treatment with purified, bacterial sphingomyelinase similarly triggers phosphatidylserine scrambling. Moreover, eryptosis following osmotic shock is blunted by the sphingomyelinase inhibitor 3,4-dichloroiso-coumarin. The stimulation of phosphatidylserine exposure is also blunted by genetic knockout of PAF receptors (PAF receptor -/- mice), and by the PAF receptor antagonist ABT491. PAF further activates Ca2+-sensitive K+ channels ( Gardos channels) in the erythrocyte cell membrane by sensitising them for the stimulating effects of cytosolic Ca2+. The signaling through PAF does, however, not necessarily require elevated cytosolic Ca2+ concentrations, and enhanced PAF levels at least partially account for Ca2+-independent, ceramide-mediated eryptosis. Chez les oiseaux il y a un noyau dans les globules rouges. Eryptosis: externalisation des phosphatidylsérine. Augmentation du calcium et du stress oxydant.")
40
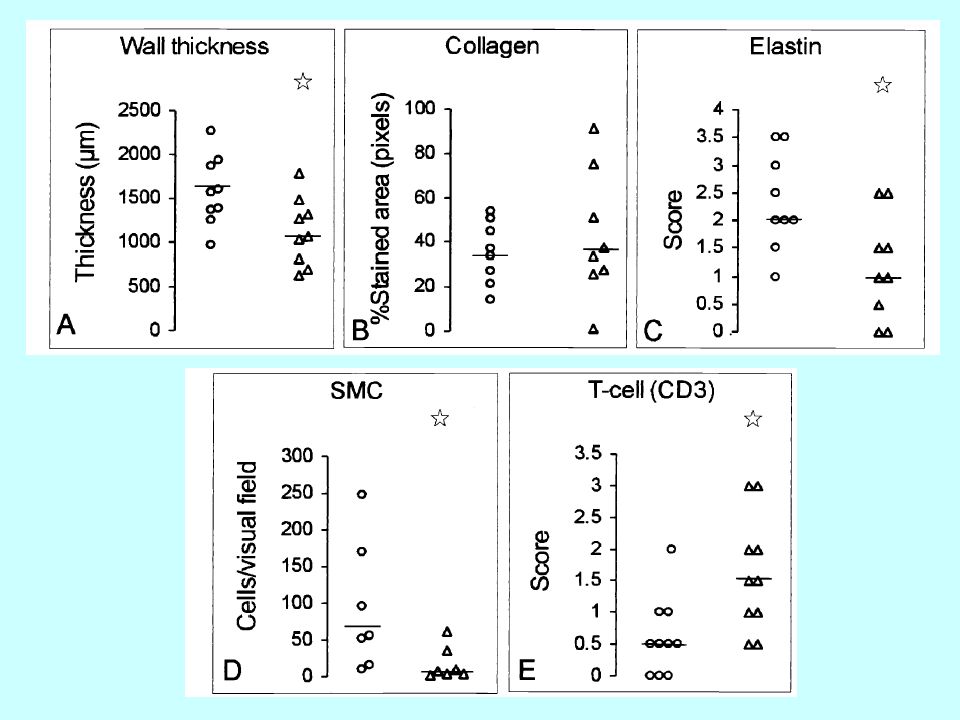
Anévrisme F F Définitions
thrombus F Définitions Anatomique : perte de parallélisme des bords de l’aorte réalisant une masse battante et expansive Physiopathologique : perte de la fonction de contention de l’artère due à une protéolyse progressive de la matrice extracellulaire F Changements dégénératifs dans la média Elastine Collagène Protéoglycanes disparition des CML

41
NT T

42
NT T

44
NT T

45
NT T

48
NT media adventice

Présentations similaires






>")













>")

