Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
L’exploration du métabolisme des acides aminés
Dr Morsli.M 4 eme année pharmacie

2
Plan Introduction Structure des acides amines
Métabolisme des acides aminés Les pathologies du métabolisme des acides amines Exploration des pathologies du métabolisme des acides amines

3
Introduction acides amines
Les acides amines constituent avec les glucides et les lipides les briques constitutives de la cellule Ils proviennent essentiellement de la dégradation des protéines alimentaires ainsi que des protéines endogènes ou bien d’une synthèse de novo acides amines Protéines endogènes Protéines alimentaires Synthèse de novo

4
Introduction La structure des acides amines leur procure des propriétés physico-chimique qui seront utiles pour leurs mise en évidence pour le diagnostic des aminoacidopathies, qui sont des pathologies dues à des déficits enzymatiques des voies du métabolisme des acides amines

5
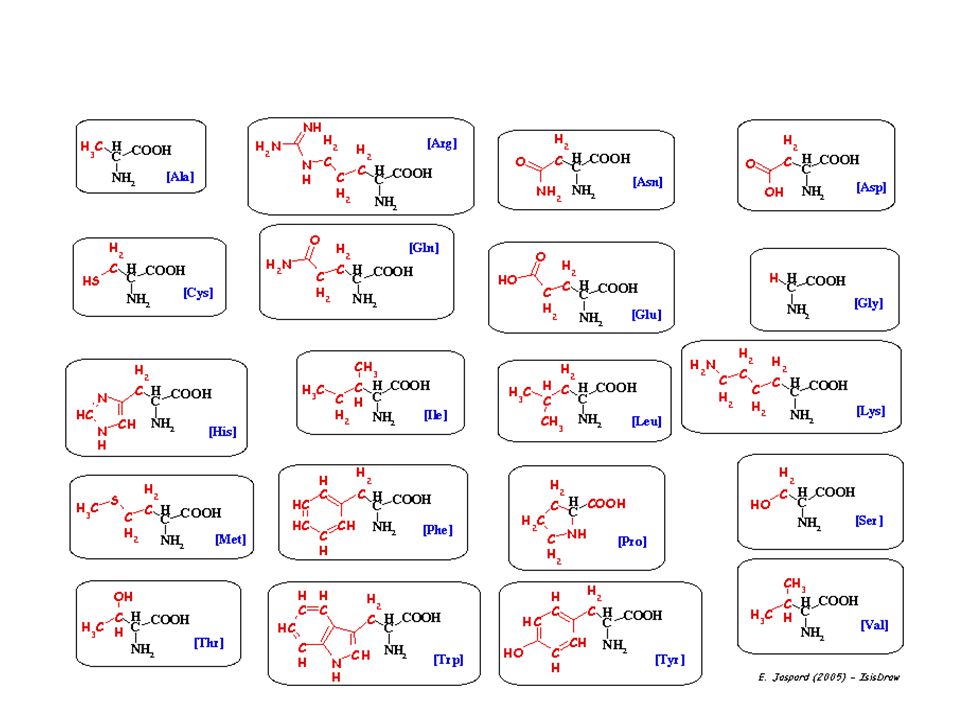
structure des acides amines
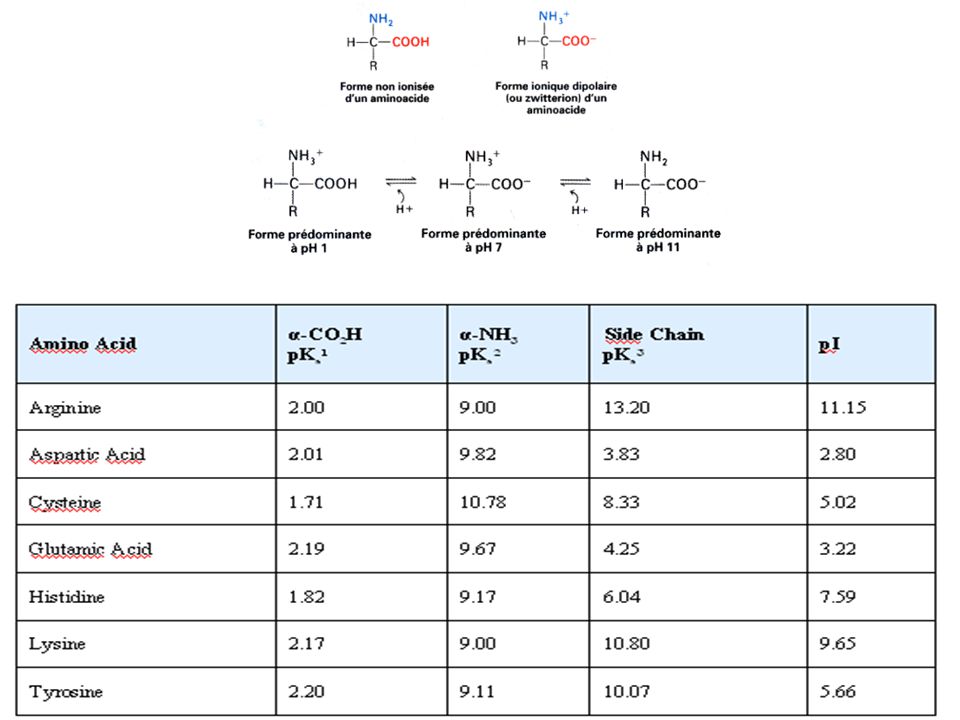
Les acides amines sont des molécules qui possèdent : une fonction acide carboxylique Une fonction amine primaire Une chaine latérale R COOH Cette structure leur procure des propriétés physico-chimiques qui sont exploitées pour leur mise en évidence, ils existent des réactions communes à tous les AA qui sont dues au groupement carboxyle et amine, et des réactions spécifiques qui sont dues à la chaine latérale R NH3 R CH

7
métabolisme des acides amines
le métabolisme des AA comprend un métabolisme commun a tous qui concerne la fonction amine et la fonction carboxyle , et un métabolisme propre a chaque AA et qui concerne la chaine latérale R Métabolisme anabolisme catabolisme Synthèse à partir d’intermédiaires métaboliques

8
catabolisme NH3 COOH Décarboxylation Molécules d’intérêt biologiques
Transamination Désamination

9
Le catabolisme de l’azote amine
NH3 Désamination Transamination Oxydative Non oxydative Uréogenèse hépatique Ammoniogenèse rénale 4/5 1/5

10
La transamination Réaction de transfert réversible de groupement aminé entre un acide aminé et un corps α cétonique, la réaction est catalysée par une aminotransférase qui utilise le phosphate de pyridoxal comme cofacteur L’un des couples est toujours le glutamate/ α cétoglutarate

11
Exp : l’alanine aminotransferase
ALANINE NH α Cétoglutarate PLP PYRUVATE GLUTAMATE NH3

12
La désamination Réaction d’élimination du groupement aminé qui transforme l’acide aminé en son corps α cétonique ,la réaction est catalysée par une désaminase La désamination oxydative: Catalysée par une glutamate déshydrogénase GLUTAMATE α CéTOGLUTARATE + NH3 La désamination non oxydative: Catalysée par une déshydratase , elle concerne les AA suivants: SERINE PYRUVATE + NH3 THREONINE CETOBUTYRATE + NH3

13
La double transamination
La première transamination: Elle est spécifique à l’AA: AA NH α CETOGLUTARATE AC α CETONIQUE GLUTAMATE NH3 La deuxième transamination : GLUTAMATE NH pyruvate α CETOGLUTARATE alanine NH3 GLUTAMATE NH OXALOACETATE α CETOGLUTARATE ASPARTATE NH3 ALAT ASAT

14
La transdésamination La transamination: catalysée par une aminotransférase AA NH α CETOGLUTARATE AC α CETONIQUE GLUTAMATE NH3 La désamination : catalysée par une glutamate déshydrogénase GLUTAMATE α CéTOGLUTARATE + NH3

15
L’ammoniogenèse Le NH3 issu de la transdésamination , réagit dans le muscle avec le glutamate pour la synthèse de glutamine, réaction catalysée par la glutamine synthétase NH3 +glutamate NH3 glutamine (2 NH3) La glutamine part à destination du rein pour subir 2 désaminations successives catalysée par une glutaminase, et libère le NH4+ qui sera éliminé dans les urines
 La glutamine part à destination du rein pour subir 2 désaminations successives catalysée par une glutaminase, et libère le NH4+ qui sera éliminé dans les urines")
16
L’uréogenèse 5 1 2 4 3 urée NH3+ HCO3- ornithine arginine
Carbamoyl phosphate 2 citrulline 4 fumarate 3 asparate argininosuccinate

17
1 Carbamoyl phosphate synthétase
2 Ornithine transcarbamylase 3 Argininosuccinate synthétase 4 Argininosuccinase 5 Arginase

18
Les pathologies du métabolisme des acides amines
Ce sont des maladies héréditaires qui se transmettent selon le mode autosomique récessif , elles sont dues à des mutations des gènes qui codent pour les enzymes des voies du métabolisme des acides amines

19
I . Les déficits génétique du cycle de l’urée
Certaines anomalies congénitales portent sur l’une des enzymes impliquées dans la formation de l’urée , ce qui a pour conséquences une hyperammoniémie car l’uréogenèse est la voie principale d’ élimination du NH3 sous forme d’urée les symptômes qui se manifestent sont causés par la toxicité de l’ammoniac pour le système nerveux central , il s’agit de nausées , vomissement , trouble du tonus et coma

20
Les déficits génétique du cycle de l’urée
PATHOLOGIE ENZYME DEFICIENTE SYMPTOMES HYPERAMMONIEMIE TYPE I CARBAMOYL PHOSPHATE SYNTHETASE LETHARGIE,CONVULSION, MORT HYPERAMMONIEMIE TYPE II ORNITHINE TRANSCARBAMYLASE ACIDEMIE ARGININOSUCCINIQUE ARGININOSUCCINASE VOMISSEMENT, CONVULSION ARGININEMIE ARGINASE RETARD MENTAL

21
L’uréogenèse 5 1 2 4 3 urée NH3+ HCO3- ornithine arginine
Carbamoyl phosphate 2 citrulline 4 fumarate 3 asparate argininosuccinate

22
II . Les phénylcétonuries
Définition et incidence: C’est une maladie génétique héréditaire qui se transmet selon le mode autosomique récessif , c’est la plus fréquente des maladies héréditaire du métabolisme des AA ( 1/10000 ) Elle est due à un déficit en phénylalanine hydroxylase PAH, ou plus rarement à son cofacteur la tetrahydrobioptérine THB par déficit de la dihydrobioptérine réductase ,l’enzyme qui permet la régénération de THB
 Elle est due à un déficit en phénylalanine hydroxylase PAH, ou plus rarement à son cofacteur la tetrahydrobioptérine THB par déficit de la dihydrobioptérine réductase ,l’enzyme qui permet la régénération de THB.")
23
DHB REDUCTASE THB DHB PHENYLALANINE TYROSINE PAH

24
b) Clinique: L’enfant est normal à la naissance , des signes autistiques apparaissent au 6eme mois qui se développent en retard mental due à l’action toxique de la phy sur la myélinisation des nerfs Une hypothyroidie par hypotyrosinémie Une hypo pigmentation par déficit en mélanine MELANINE PHENYLALANINE TYROSINE THYROXINE CATECHOLAMINE

25
c) Biologie: Le déficit en PAH entraine une accumulation de la phy dans le sang , ce qui entraine l’activation d’une voie mineur dans les conditions physiologique qui synthétise les corps cétoniques de la phy qui sont éliminés dans les urines d’où le nom phenylcétonurie PHENYLALANINE PHENYLPYRUVATE PHENYLLACTATE PHENYLACETATE

26
D) Traitement: Il s’agit d’un régime pauvre en phy, car cette AA reste essentielle à l’organisme Une concentration élevée en phy chez la femme enceinte phenylcétonurique peut provoquer des anomalies du développement du fœtus Dans les pays développés le dépistage de la PCU est systématique à la naissance

27
III Les erreurs innées du métabolisme de la tyrosine
TYROSINE AMINOTRANSFERASE TYROSINEMIE II 4-OH PHENYLPYRUVATE 4OH PHENYLPYRUVATE OXYGENASE TYROSINEMIE III HOMOGENTISATE HOMOGENTISATE OXYGENASE ALCAPTONURIE MALEYLACETOACETATE FUMARYL ACETOACETASE FUMARYLACETOACETATE TYROSINEMIE I FUMARATE ACETOACETATE

28
La tyrosinémie type II:
Due à un déficit en fumarylacetoacetase, ce qui provoque l’accumulation de fumarylacetoacetate qui est à l’origine d’atteintes hépatiques conduisant à des cirrhose , et d’atteintes tubulaires conduisant au syndrome de Fanconi La tyrosinémie type II: Due à un déficit en tyrosine aminotransferase, peut affecter les yeux, la peau et le développement mental. Les premiers symptômes sont le larmoiement excessif, la douleur oculaire et l'apparition d'ulcères à la corné. Une déficience intellectuelle est notée sur 50 % des personnes atteintes. La tyrosinémie typeIII: Due à un déficit en 4 OH phenylpyruvate dioxygenase. Elle cause des convulsions, une déficience intellectuelle et, de façon intermittente, une absence de coordination des membres (ataxie).
.")
29
L’alcaptonurie: L’albinisme:
Due à un déficit en homogentisate dioxygenase qui provoque l’accumulation d’acide homogentisique donnant à la peau une coloration brunâtre, c’est une affection bénigne L’acide homogentisique est incolore mais s’oxyde à l’air libre donnant une coloration brunâtre aux urines L’albinisme: Due à un déficit en tyrosinase , l’enzyme qui permet la synthèse de mélanine à partir de tyrosine ce qui induit une perte de pigmentation ( cheveux blanc , peau rose ) TYROSINASE TYROSINE MELANINE
 TYROSINASE. TYROSINE. MELANINE.")
30
IV La leucinose: C’est un déficit enzymatique en déshydrogénase des corps cétoniques des acides amines à chaine ramifiée, ce qui provoque l’accumulation de valine, leucine et isoleucine qui seront éliminés dans les urines et lui confèrent une odeur de sirop d’érable Les signes cliniques sont des vomissements, convulsion et mort précoce

31
V. Les hyperhomocysteinémies
Maladie héréditaire transmise selon le mode autosomique récessif, elle est due à un trouble enzymatique du métabolisme de la méthionine .Le défaut enzymatique porte sur la cystathionine-synthéase. METHIONINE HOMOCYSTEINE cystathionine-synthéase. CYSTATHIONINE

32
Le taux d'homocysteine sanguin est très augmenté (100 micromole / L au lieu de 10 micromole normalement). Cette hyperhomocysteinémie est responsable d’ accidents vasculaires, thromboemboliques ou plus rarement d’athérosclérose. L’homocystéine a un rôle délétère sur l’endothélium vasculaire, et stimulent la production des médiateurs de l’inflammation

33
Tab :les maladies génétiques affectant le métabolisme des acides aminés
LES PATHOLOGIES ENZYME DEFICIENTE SYMPTOMES BIOLOGIE HYPERAMMONIEMIE TYPE I CARBAMOYL PHOSPHATE SYNTHETASE LETHARGIE,CONVULSION, MORT AMMONIUM HYPERAMMONIEMIE TYPE II ORNITHINE TRANSCARBAMYLASE ACIDEMIE ARGININOSUCCINIQUE ARGININOSUCCINASE VOMISSEMENT, CONVULSION AMMONIUM , ARGININOSUCCINATE ARGININEMIE ARGINASE RETARD MENTAL AMMONIUM, ARGININE PHENYLCETONURIE PHY HYDROXYLASE DHB REDUCTASE RETARD MENTAL, HYPOPIGMENTATION PHY, PHENYLACETATE, PHENYLLACTATE, TYROSINE TYROSINEMIE I FUMARYLACETOACETASE ATTEINTES HEPATIQUE ET TUBULAIRE FUMARYLACETOACETATE ET MALEYLACETOACETATE ALCAPTONURIE HOMOGENTISATE DIOXYGENASE PIGMENTS NOIR SUR LA PEAU HOMOGENTISATE LEUCINOSE DESHYDROGENASE DES CORPS CETONIQUES DES AA RAMIFIEE CONVULSION LEUCINE,VALINE ET ISOLEUCINE HYPERHOMOCYSTEINEMIE CYSTATHIONINE SYNTHETASE ACCIDENTS THROMBOEMBOLIQUES HOMOCYSTEINE SANGUIN ET URINAIRE

34
Exploration des pathologies du métabolisme des acides amines
L’exploration repose sur la structure chimique des acides aminés qui procure une réactivité aux différents groupements fonctionnelles et permet leurs séparation , identification et quantification dans un milieu biologique

35
EXPLORATION DOSAGE SPECIFIQUE DEPISTAGE QUANTIFICATION FRACTIONNEMENT

36
I. Techniques de dépistage:
Ce sont des réactions qui permettent de détecter rapidement une concentration anormale en AA ou en leur catabolites, elles se font sur les urines fraiches du matin Réaction de BRAND: permet de mettre en évidence les AA soufrés comme la cysteine et l’homocysteine, qui développent une coloration rouge avec le nitroprussiate de sodium en milieu alcalin Le 2,4 dinitrophenylhydrazine DNPH en milieu chlorhydrique donne avec les acides cétoniques un précipité jaune d’hydrazone Le perchlorure de fer fecl 3 en milieu acide donne une coloration variable selon le dérivé PHENYLPYRUVATE VERT ± FUGACE TYROSINE ET DERIVE VERT FUGACE DERIVE DE L’HISTIDINE BLEU VERT STABLE DERIVE DE LA LEUCINE BLEU

37
4) Les ions mercuriques :
En présence de nitrites , la tyrosine et ses métabolites donnent un composé rouge violacée NB: Le dépistage permet d’orienter le diagnostic

38
II. Techniques de quantification des acides amines
Réaction à la ninhydrine: Les AA en présence de ninhydrine,à chaud et en milieu tamponné à Ph 5,5 forment un composé violacé appelé pourpre de ruhemann dont l’absorbance est mesuré à 570nm Avec la proline qui est un iminoacide ont obtient une coloration jaune qui absorbe à 440nm Réaction à la fluorescamine: La fluorescamine réagit rapidement avec les AA dans un tampon borate PH9 et permet d’obtenir un dérivé fluorescent très sensible qui détecte des concentration de l’ordre du nano gramme Le chlorure de dansyle: Il forme avec les AA un dansylaminoacide fluorescent Le 1 fluoro 2,4 dinitrobenzene DNFB ( réaction de Sanger ) À ph 9 les AA forment avec le DNFB le dinitrophenylaminoacide coloré en jaune qui absorbe à 420 nm Rmq: Ces réactifs réagissent avec le groupement α aminé des AA
 À ph 9 les AA forment avec le DNFB le dinitrophenylaminoacide coloré en jaune qui absorbe à 420 nm. Rmq: Ces réactifs réagissent avec le groupement α aminé des AA.")
39
III. Technique de fractionnement des acides amines
Chromatographie sur couche mince: Cette technique permet la séparation des AA entre 2 phases: Phase stationnaire :la couche de gel en silice ou cellulose Phase mobile: un solvant qui est un mélange de butanol, d’acide acétique et d’eau Après migration des AA sur le gel , la révélation se fait à la ninhydrine, on peut identifier l’acide aminé grâce à la comparaison avec des témoins qui sont des standards d’acides aminés

40
III. Techniques de fractionnement des acides amines
Electrophorèse sur papier: Les AA soumis à un tampon acide acquirent une charge qui dépend du groupement fonctionnelle R, cette différence de charge permet leurs séparation par migration lorsqu'ils sont soumis à un champ électrique Electrochromatographie: C’est un couplage entre électrophorèse dans une première étape, et une chromatographie dans une étape ultérieur Cette technique a un excellent pouvoir de résolution

41
IV. Techniques de dosage spécifique
Méthodes chromatographiques Méthodes chimiques Méthodes microbiologiques

42
a) Méthodes chromatographiques :
La chromatographie échangeuse d’ions: C’est une technique qui permet la séparation, l’identification et la quantification de chaque AA dans un liquide biologique La colonne de chromatographie est formé d’un tube remplie de particules synthétiques chargées , ceux avec groupements anioniques sont appelés résines échangeuses de cations et ceux qui possèdent des groupements cationiques sont appelés résines échangeuses d’anions Les AA seront élués en fonction de leur affinité pour la résine

43
Exp: colonne échangeuse de cations
Des groupements anioniques sont fixés sur la résine SO3- qui attire les AA chargés positivement La solution d’acide aminé est amené à ph 3, à ce ph tous les AA sont chargés positivement, cette charge diffère par le radical R Les AA ayant la charge la plus positive se lie solidement à la résine et migrent plus lentement ,ceux ayant la charge la moins positive migrent rapidement et seront élués les premiers. Chaque fraction recueillie à la sortie de la colonne est quantifiées par la réaction à la ninhydrine

45
a)Méthodes chromatographiques
La chromatographie liquide haute performance HPLC : C’est une chromatographie à haute pression , la pression permet une séparation en un temps plus court avec une meilleur résolution, l’élution, la collection et l’analyse des fractions sont automatisés , le résultat est un chromatographe qui donne la nature de l’AA par le temps de rétention et sa concentration par la surface du pic . C’est la technique de référence pour l’étude des AA

46
b) Méthodes microbiologiques:
Test de Guthrie: La phenylalanine est un facteur essentiel à la croissance de certaines souches de Bacillus subtilus On additionne à une gélose le B2 thyenylalanine ,c’est un antagoniste de la phy qui inhibe la croissance bactérienne On étale sur la gélose une suspension de la bactérie et on pose un disque de papier imprégné de sang , si la concentration de la phy dépasse 120 umol/l on aura une croissance bactérienne , la zone de croissance est comparée avec celle obtenues à l’aide de disques témoins

47
c) Méthodes chimiques:
Ces méthodes permettent de doser spécifiquement des AA ou des produits de leur métabolisme Dosage de la phenylalanine: réaction de McCaman et Robins En présence d’un dipeptide L-leucyl-L-alanine, la PHY forme un complexe fluorescent avec la ninhydrine-Cu Dosage de la tyrosine: Le 1 nitroso 2 naphtol et le nitrite forment en solution d’acide nitrique avec la tyrosine un complexe fluorescent Dosage de l’ammoniac: αCetoglutarate+NADH,H +NH NAD+glutamate On mesure la diminution de l’absorbance du NADH,H à 340 nm Glutamate déshydrogénase

48
Concentrations plasmatiques des acides amines et dérivés
AMINOACIDES ET DERIVES CONCENTRATION PLASMATIQUE µmol/l ORNITHINE ARGININE VALINE LEUCINE ISOLEUCINE PHENYLALANINE TYROSINE Concentrations plasmatiques des acides amines et dérivés

Présentations similaires








 Obtention de l’ADN recombinant>")










