Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Algorithmes de filtrage
Nadia El-Mabrouk

2
Plan Introduction Objectif Qu’est-ce qu’une heuristique? Algorithmes de filtrage: Principe et méthode exacte (Baeza-Yates-Perlberg, 1992) Heuristique FASTA Optimisation possible en introduisant le voisinage d’un k-mer Heuristique BLAST Ce qui est derrière BLAST: Comment choisir la taille k d’un k-mer? Comment étendre les k-mers en temps sous linéaire? Graines espassées. Méthode de PatternHunter Alignement de génomes entiers

3
1. Introduction Recherche de P de taille m dans T de taille n à d erreurs près Programmation dynamique: Temps O(mn) Différentes améliorations: Temps O(dn) Est-ce qu’on peut faire mieux? Temps sous-linéaire?
 Différentes améliorations: Temps O(dn) Est-ce qu’on peut faire mieux Temps sous-linéaire")
4
Heuristique ou méthode exacte?
Heuristique pour la recherche d’un motif: méthode permettant de trouver la « plupart » des occurrences, mais pas de garantie de les trouver toutes, et peut se tromper. Faux-négatifs: Occurrences non détectées. Faux-positifs: Motifs trouvés qui ne sont pas des occurrences:

5
Heuristique Sélectivité (spécificité): Capacité à ne détecter que la réalité biologique et rien de plus. Problème des Faux-Positifs Sensibilité: Capacité à détecter tout ce qui est intéressant sur le plan biologique. Problème des Faux-Négatifs

6
2. Algorithmes de filtrage
Effectuer un premier passage sur T pour éliminer toutes les parties qui ne sont pas susceptibles de contenir P. Partitionner P (ou T) en facteurs de taille k (k-mers, seeds ou graines). Construire un index de ces facteurs. Phase de recherche: Utiliser une méthode de recherche exacte pour trouver toutes les occurrences de ces facteurs dans T, en temps (sous) linéaire Phase de vérification: Utiliser une méthode de recherche approchée dans un intervalle restreint autour de chaque facteur trouvé, en temps (sous) linéaire.
 en facteurs de taille k (k-mers, seeds ou graines). Construire un index de ces facteurs. Phase de recherche: Utiliser une méthode de recherche exacte pour trouver toutes les occurrences de ces facteurs dans T, en temps (sous) linéaire. Phase de vérification: Utiliser une méthode de recherche approchée dans un intervalle restreint autour de chaque facteur trouvé, en temps (sous) linéaire.")
7
Algorithmes de filtrage
Les k-mers définissent des ancrages dans la table de programmation dynamique P = p1p2p3p4 T p1 p2 P p3 p4

8
Algorithmes de filtrage
Les k-mers définissent des ancrages dans la table de programmation dynamique P = p1p2p3p4 T p1 p2 P p3 p4 +/-d

9
Une méthode exacte (Baeza-Yates-Perlberg, 1992)
Partition de P en régions de taille k = ENT(m/d+1) d+1 régions de taille k, plus au plus une région de taille < k. Si le facteur T’ de T est une occurrence de P à d erreurs près, alors il existe au moins une région R de P et un facteur de même taille T’’ de T’ tel que R et T’’ coïncident exactement. T’ T’’ T: P: R
 d+1 régions de taille k, plus au plus une région de taille < k. Si le facteur T’ de T est une occurrence de P à d erreurs près, alors il existe au moins une région R de P et un facteur de même taille T’’ de T’ tel que R et T’’ coïncident exactement. T’ T’’ T: P: R.")
10
Idée générale (Baeza-Yates-Perlberg, 1992)
P: Ensemble des d+1 premières régions de P. Construire un « index » de P. Trouver l’ensemble I des pos. des occurrences de P dans T. Étendre les occurrences par programmation dynamique. Construction d’un index possible en temps et espace O(m): Arbre des préfixes (Aho-Corasick), « trie structure». Recherche exacte possible en O(n) Phase de vérification: O(hdm) où h=|I|. O(m+n+hdm)
: Arbre des préfixes (Aho-Corasick), « trie structure». Recherche exacte possible en O(n) Phase de vérification: O(hdm) où h=|I|. O(m+n+hdm)")
11
3. Heuristique FASTA (Lipman, Pearson 1985)
Taille de graine k fixée (en général 6 pour nuc.2 pourAA) Indexer tous les facteurs de taille k de P (O(m)) Rechercher toutes les occurrences exactes de ces facteurs de P dans T (O(m+n)) Chainer les graines pour former des alignements complets ~ temps proportionnel au nombre h de graines
 Indexer tous les facteurs de taille k de P (O(m)) Rechercher toutes les occurrences exactes de ces facteurs de P dans T (O(m+n)) Chainer les graines pour former des alignements complets. ~ temps proportionnel au nombre h de graines.")
12
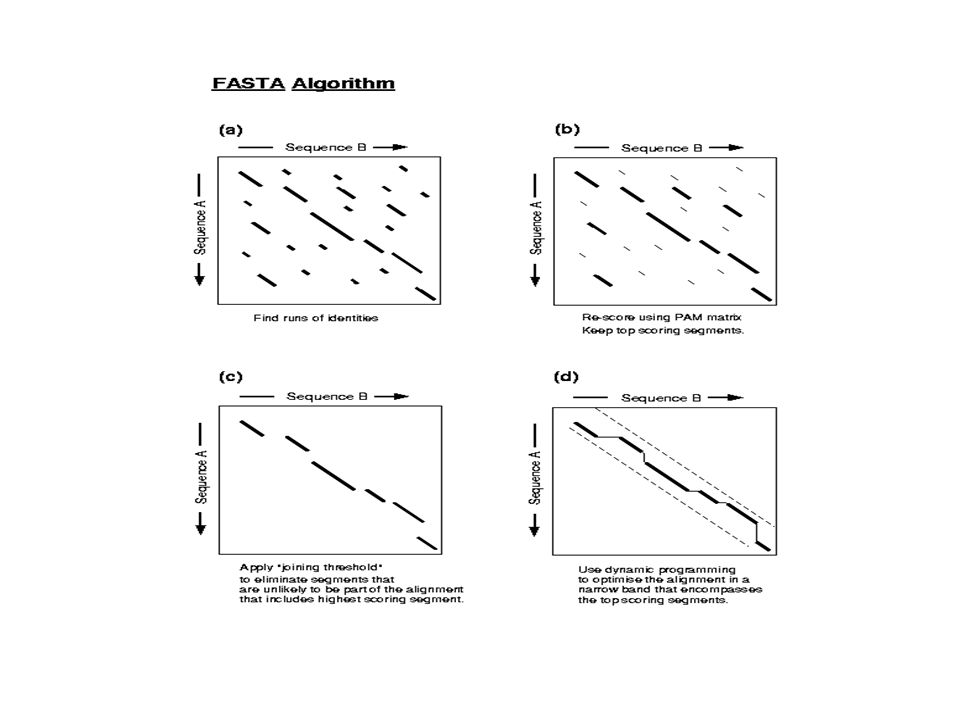
Heuristique FASTA (Lipman, Pearson 1985)
Pour une valeur ktup donnée (en général 6 pour nuc.2 pourAA), trouver toutes les paires de séquences de taille ktup identiques dans P et T: hot-spot Déterminer des zones denses en identité: hot-spot consécutifs sur chaque diagonale. Score d’une zone: Score positif pour chaque hot-spot Score négatif pour les espaces entre les hot-spot FASTA garde les 10 zones de score optimal. Zones contenant des matchs et mismatchs
, trouver toutes les paires de séquences de taille ktup identiques dans P et T: hot-spot. Déterminer des zones denses en identité: hot-spot consécutifs sur chaque diagonale. Score d’une zone: Score positif pour chaque hot-spot. Score négatif pour les espaces entre les hot-spot. FASTA garde les 10 zones de score optimal. Zones contenant des matchs et mismatchs.")
13
Réaligner chaque zone, en considérant une matrice de substitution (PAM ou BLOSUM)
Init1: Meilleur alignement obtenu Parmi les 10 zones, garder celles dont le score dépasse un seuil ``cut-off’’. Combiner les zones en une seule Initn: Contient insertions/suppressions/mismatchs Programmation dynamique dans une bande autour de Init1 (bande de taille 16 si ktup=2) Opt: Meilleur alignement obtenu Au cours de la recherche, statistiques calculées pour Init1, Initn, Opt: alignements significatifs ou non.
 Opt: Meilleur alignement obtenu. Au cours de la recherche, statistiques calculées pour Init1, Initn, Opt: alignements significatifs ou non.")
15
Taille des graines? Complexité de l’heuristique de filtrage dominée par h: nombre de graines. Combien de graines attendues pour deux séquences aléatoires sur un alphabet S? Un mot de taille k a une probabilité de 1/|S|k d’apparition dans l’une ou l’autre des séquences. h de l’ordre de nm |S|-k (4-knm dans le cas de séquences d’ADN) Complexité totale de l’heuristique de filtrage O(n+m+ |S|-kndm2) D’autant plus efficace que k est grand Mais plus k est grand plus on a de chance de manquer des occurrences.
 Complexité totale de l’heuristique de filtrage O(n+m+ |S|-kndm2) D’autant plus efficace que k est grand. Mais plus k est grand plus on a de chance de manquer des occurrences.")
16
4. Voisinage d’un mot Plutôt que de rechercher les occurrences exactes des k-mers, rechercher un « voisinage » des k-mers: mots qui sont à moins de e% d’erreurs, ou à une distance ≤ d (d = e m) Nd(w) = { v / v et w ont au plus d différences} N1(abbaa) = { aabaa, aabbaa, abaa, abaaa, ababaa, abba, abbaa, abbab, abbaaa, abbaba, abbba, abbbaa, babbaa, bbaa, bbbaa} (1-match ou 20%-match)
 Nd(w) = { v / v et w ont au plus d différences} N1(abbaa) = { aabaa, aabbaa, abaa, abaaa, ababaa, abba, abbaa, abbab, abbaaa, abbaba, abbba, abbbaa, babbaa, bbaa, bbbaa} (1-match ou 20%-match)")
17
Puissance du voisinage
Supposons que l’on cherche un 3-match d’un mot P de taille 40. Si on choisit k=4 et qu’on divise P en 4 10-mers, alors au moins un doit matcher exactement. => Occurrence tous les |S|10/4 caractères (e.g. 2.5 ⋅ 105 pour les nucléotides) T: P:
 T: P:")
18
Puissance du voisinage
Supposons que l’on cherche un match un 3-match d’un mot P de taille 40. Si on divise le mot requête en 2 20-mers, alors au moins un 1-voisin des deux doit matcher. => Occurrence tous les |S|20/2|N1(20)| caractères (e.g / 2 ⋅ 160 = 3.12 ⋅ 109 pour les nuc.) T: N1 (P): 10,000 fois plus spécifique ! (mais beaucoup plus de mots pour la recherche exacte)
| caractères (e.g / 2 ⋅ 160 = 3.12 ⋅ 109 pour les nuc.) T: N1 (P): 10,000 fois plus spécifique ! (mais beaucoup plus de. mots pour la recherche exacte)")
19
5. BLAST Recherche exacte sur un voisinage des k-mers
Distance Hamming pondérée (e.g. PAM120) Extension: stop quand le score chute sous une valeur seuil. BLAST est une heuristique “BLAST” inspiré de “SLAM” = SubLinear Approximate Match
 Extension: stop quand le score chute sous une valeur seuil. BLAST est une heuristique. BLAST inspiré de. SLAM = SubLinear Approximate Match.")
20
BLAST Exemple: Pour w =PQG, {PQG, PRG, PKG, PDG, PMG…}
Former la liste de tous les k-mers (seeds ou graines) de la séquence requête P P Maximum l-k+1 mots Pour chaque facteur w, former la liste de tous les mots de taille k dont le score avec w dépasse un seuil s Exemple: Pour w =PQG, {PQG, PRG, PKG, PDG, PMG…}
 de la séquence requête P. P. Maximum l-k+1 mots. Pour chaque facteur w, former la liste de tous les mots de taille k dont le score avec w dépasse un seuil s. Exemple: Pour w =PQG, {PQG, PRG, PKG, PDG, PMG…}")
21
Identifier les occurrences exactes des mots de la liste dans la BD
Pour chaque paire de séquences trouvées, étendre l’alignement dans les deux directions, jusqu’à ce que le score de l’alignement chute de X par rapport à sa valeur d’origine. Segment accepté si score>S

22
Le HSP de score maximal sur l’ensemble de la séquence est appelé Maximal Scoring segment Pair (MSP)
Les alignements locaux HSP sont chaînés pour former des alignements plus longs, incluant des espaces et des trous. Si le MSP ou les HSP combinés ont un score qui dépasse un certain seuil S, il sont affichés

23
Paramètres La séquence format FASTA La banque (compressée)
k (taille du mot). Protéines: k de 3 à 5, et s = 17 Donne à peu près 50 mots pour chaque facteur Nucléotides: k = 11 ou 12 S (seuil de sélection d’un score) Matrices de substitution (BLOSUM 62) ou score pour les nucléotides (+5/-4)
. Protéines: k de 3 à 5, et s = 17. Donne à peu près 50 mots pour chaque facteur. Nucléotides: k = 11 ou 12. S (seuil de sélection d’un score) Matrices de substitution (BLOSUM 62) ou score pour les nucléotides (+5/-4)")
24
Évaluation statistique
Expect-value = nb de fois où un HSP est attendu par chance sur l’ensemble de la banque. Plus cette valeur est faible, plus le HSP est significatif P-value: P(N): Probabilité du score observé. Plus cette valeur est faible, plus le HSP est significatif.
: Probabilité du score observé. Plus cette valeur est faible, plus le HSP est significatif.")
25
Plan Introduction Objectif Qu’est-ce qu’une heuristique? Algorithmes de filtrage: Principe et méthode exacte (Baeza-Yates-Perlberg, 1992) Heuristique FASTA Optimisation possible en introduisant le voisinage d’un k-mer Heuristique BLAST Ce qui est derrière BLAST: Comment choisir la taille k d’un k-mer? Comment étendre les k-mers en temps sous linéaire? Graines espassées. Méthode de PatternHunter Alignement de génomes entiers

27
Taille de la graine? The Splitting Lemma
Lemme: Si w= w0 w1 match v à moin de e% erreurs, alors: (a) w0 match un préfixe v0 de v à moins de e% erreurs, ou (b) w1 match un suffixe v1 de v à moins de e% erreurs. W d erreurs d= e |W| v W0 W1 W ≤ Ld/2˩erreurs? ≤ Ld/2˩ erreurs? v W0 W1 ≤ Ld/2˩ erreurs V1
 w0 match un préfixe v0 de v à moins de e% erreurs, ou. (b) w1 match un suffixe v1 de v à moins de e% erreurs. W. d erreurs. d= e |W| v. W0. W1. W. ≤ Ld/2˩erreurs ≤ Ld/2˩ erreurs v. W0. W1. ≤ Ld/2˩ erreurs. V1.")
28
W0 W1 ≤ Ld/2˩ erreurs W V1 W0 W1 W11 W10 ≤ Ld/4˩? ≤ Ld/4˩? W V10 W0 W1 W11 W10 W100 W101 ≤ Ld/8˩? ≤ Ld/8˩? V101

29
Taille de la graine? T P Prendre k=log|S| n comme taille de graine.
Lemme: Tout e%-match de P a un e%-match à au moins un seed de taille log|S| n T e%-match P

30
Phase de vérification? T P Prendre k=log|S| n comme taille de graine.
Temps moyen pour la vérification: O(h.dlog|S| n) où h est le nombre d’oc.des graines T P
 où h est le nombre d’oc.des graines. T. P.")
31
Supposons graines de taille 3
e = 25 . Pour P de taille 12, d=3 . Supposons graines de taille 3 P : g t a c P/T a t c g

32
7. Graines espacées BLAST trouve une graine de taille 11 qui match, puis étend GCNTACACGTCACCATCTGTGCCACCACNCATGTCTCTAGTGATCCCTCATAAGTTCCAACAAAGTTTGC || ||||| | ||| |||| || |||||||||||||||||| | |||||||| | | ||||| GCCTACACACCGCCAGTTGTG-TTCCTGCTATGTCTCTAGTGATCCCTGAAAAGTTCCAGCGTATTTTGC GAGTACTCAACACCAACATTGATGGGCAATGGAAAATAGCCTTCGCCATCACACCATTAAGGGTGA---- || ||||||||| |||||| | ||||| |||||||| ||| |||||||| | | | || GAATACTCAACAGCAACATCAACGGGCAGCAGAAAATAGGCTTTGCCATCACTGCCATTAAGGATGTGGG TGTTGAGGAAAGCAGACATTGACCTCACCGAGAGGGCAGGCGAGCTCAGGTA ||||||||||||| ||| ||||||||||| || ||||||| || |||| | TTGACAGTACACTCATAGTGTTGAGGAAAGCTGACGTTGACCTCACCAAGTGGGCAGGAGAACTCACTGA GGATGAGGTGGAGCATATGATCACCATCATACAGAACTCAC CAAGATTCCAGACTGGTTCTTG ||||||| |||| | | |||| ||||| || ||||| || |||||| ||||||||||||||| GGATGAGATGGAACGTGTGATGACCATTATGCAGAATCCATGCCAGTACAAGATCCCAGACTGGTTCTTG

33
Exemple d’une occurrence manquée (Exemple de B. Ma)
Pas de graine de taille 11 qui match, pourtant similarité de 80%: GAGTACTCAACACCAACATTAGTGGGCAATGGAAAAT || ||||||||| |||||| | |||||| |||||| GAATACTCAACAGCAACATCAATGGGCAGCAGAAAAT Dilemme: Sensibilité – nécessite des graines courtes Capacité à détecter les homologies Rapidité – nécessite des graines plus longues Mega-BLAST utilise des graines de taille 28.

34
PatternHunter utilise des “graines espacées”
(appelé modèle) 11 matchs requis (poids=11) 7 positions “don’t care” GAGTACTCAACACCAACATTAGTGGCAATGGAAAAT… || ||||||||| ||||| || ||||| |||||| GAATACTCAACAGCAACACTAATGGCAGCAGAAAAT… Hit = Tous les matchs requis sont satisfaits Modèle de BLAST =
 11 matchs requis (poids=11) 7 positions don’t care GAGTACTCAACACCAACATTAGTGGCAATGGAAAAT… || ||||||||| ||||| || ||||| |||||| GAATACTCAACAGCAACACTAATGGCAGCAGAAAAT… Hit = Tous les matchs requis sont satisfaits. Modèle de BLAST =")
35
VI. Simulated sensitivity curves

36
Pourquoi sensibilité meilleure?
Les copies ‘shiftées’ des graines espacées ne chevauchent pas trop: ...... Les ‘Hits’ à différentes positions sont plus indépendants Plus les copies shiftées sont indépendantes, plus on augmente la probabilité d’identifier une homologie. Moins il y a de similarités entre deux copies shiftées, plus le modèle est susceptible de donner une bonne sensibilité.

37
VI. Pourquoi plus rapide avec des graines espacées?
CAA?A??A?C??TA?TGG? |||?|??|?|??||?|||? TTGACCTCACC? |||||||||||? Une homologie donne lieu à plusieurs ‘hits’ par BLAST (redondance) Graines espacées donnent lieu à moins de ‘hits’ pour chaque homologie
 Graines espacées donnent lieu à moins de ‘hits’ pour chaque homologie.")
38
``Indeed, under the condition that there is one hit in a length 64, 70% similar homology, the average number of hits in that region is 2.0 for PH’s weight-11 seed 3.6 for contiguous weight-11 seed.’’

39
Observations (B. Ma) Des modèles différents peuvent détecter différentes homologies Deux conséquences: Certains modèles sont meilleurs que d’autres On peut utiliser simultanément plusieurs modèles de graines Approcher les 100% de sensibilité. PatternHunter II

40
8. Alignement de génomes entiers
Comparaison de génomes entiers permet de: Identifier les séquences codantes dans les 2 espèces Localiser les facteurs de transcription et les signaux de régulation Comprendre les mécanismes et l’histoire de l’évolution génomique Similarités et différences dans l’ordre des gènes Smith-Waterman, et même FASTA ou BLAST trop lents et pas adaptés à la comparaison de génomes entiers. Améliorer sensibilité et temps de calcul, sans empirer la sélectivité

41
Alignement de l’homme et de la souris par BLASTZ (Schwartz et al. 2003)
Supprimer les répétitions propre à chaque espèce Trouver tous les 12-mers espacés identiques, à une ``transition’’ près, dans les deux génomes. Étendre chaque paire de 12-mers dans les deux directions (sans gaps), jusqu’à ce que le score chute en dessous d’un certain seuil Si l’alignement (sans gaps) trouvé dépasse un seuil (disons 300) Étendre l’alignement en autorisant les gaps (programmation dyn.) Garder l’alignement si le score dépasse un seuil (disons 5000) Entre chaque paire d’alignements, refaire l’étape 2. avec des scores moins contraignants. Par exemple, 7-mers (match exact), seuils plus faibles (par exemple 2000 avec et sans gaps) Rétablir les vraies positions des alignements trouvés (étape 1.)
, jusqu’à ce que le score chute en dessous d’un certain seuil. Si l’alignement (sans gaps) trouvé dépasse un seuil (disons 300) Étendre l’alignement en autorisant les gaps (programmation dyn.) Garder l’alignement si le score dépasse un seuil (disons 5000) Entre chaque paire d’alignements, refaire l’étape 2. avec des scores moins contraignants. Par exemple, 7-mers (match exact), seuils plus faibles (par exemple 2000 avec et sans gaps) Rétablir les vraies positions des alignements trouvés (étape 1.)")
42
Paramètres utilisés Matrice de substitution: Gap de taille k pénalisé
91 -114 -31 -123 100 -125 -100 Matrice de substitution: Gap de taille k pénalisé par un poids de k Score d’un alignement multiplié par une valeur entre 0 et 1 en fonction de la nature des séquences (biais des nucléotides) Les seuils doivent être très élevés pour atteindre une spécificité raisonnable (au moins 3000 pour les alignements avant gap) 12-mers espacé (19 positions): (Ma et.al 2002) Autoriser une transition: (A-G, G-A, C-T, T-C)
 Les seuils doivent être très élevés pour atteindre une spécificité raisonnable (au moins 3000 pour les alignements avant gap) 12-mers espacé (19 positions): (Ma et.al 2002) Autoriser une transition: (A-G, G-A, C-T, T-C)")
Présentations similaires