Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Introduction, présentation de la plateforme
South Green. SupAgro, Montpellier, 04 février 2013

2
Le déluge de données NGS
Next-generation sequencing

3
Rappel: synthèse de l’ADN
5’ 3’ Juste pour rappel: L’ADN polymérase (naturellement ou in vitro) est capable d’ajouter un désoxyribonucléotide à l’extrémité 3’-OH d’un brin amorce, libérant ainsi un pyrophosphate et un ion H+. Le dNTP incorporé possède une base complémentaire à celle présente sur le brin matrice. Si l’on a modifié l’extrémité 3’-OH du nucléotide incorporé, alors la synthèse du nouveau brin s’arrête. Cette modification peut être réversible ou non. De la même façon, on peut rajouter un fluorophore au niveau des phosphates: ce fluorophore sera éliminé après incorporation du dNTP. + H+ Synthèse catalysée par une ADN polymérase
 est capable d’ajouter un désoxyribonucléotide à l’extrémité 3’-OH d’un brin amorce, libérant ainsi un pyrophosphate et un ion H+. Le dNTP incorporé possède une base complémentaire à celle présente sur le brin matrice. Si l’on a modifié l’extrémité 3’-OH du nucléotide incorporé, alors la synthèse du nouveau brin s’arrête. Cette modification peut être réversible ou non. De la même façon, on peut rajouter un fluorophore au niveau des phosphates: ce fluorophore sera éliminé après incorporation du dNTP. + H+ Synthèse catalysée par une ADN polymérase.")
4
Rappel: Méthode de Sanger
-Préparation d’une grande quantité d’ADN (~1mg) -Dénaturation et ajoût de l’amorce, des 4 dNTP (dont 1 radioactif, S35 ou P32) et de l’ADN polymérase -Faire 4 aliquots et mettre un peu de ddNTP dans chaque tube -Elongation et terminaison par incorporation de didéoxynucléotides spécifiques Brin d’ADN matrice Brin d’ADN nouvellement synthétisé La méthode originale de séquençage de Sanger et al (1977) La technique est la suivante : -Une amorce radiomarquée vient s’hybrider sur le fragment ADN à séquencer Des désoxyribonucléotides (dNTPs) pour les quatre bases sont ajoutés au mélange réactionnel ainsi qu’une Polymérase. Le mélange est divisé en quatre (un par base) et on rajoute chaque fois le didésoxyribonucléotide correspondant au mélange A, T, C et G. Chaque fois qu’un ddNTP se fixe pendant la réaction d’amplification, il bloque l’ajout d’une nouvelle base. Amorce 1 ddNTP est statistiquement incorporé à toutes les positions possibles
 -Dénaturation et ajoût de l’amorce, des 4 dNTP (dont 1 radioactif, S35 ou P32) et de l’ADN polymérase -Faire 4 aliquots et mettre un peu de ddNTP dans chaque tube -Elongation et terminaison par incorporation de didéoxynucléotides spécifiques. Brin d’ADN matrice. Brin d’ADN nouvellement. synthétisé. La méthode originale de séquençage de Sanger et al (1977) La technique est la suivante : -Une amorce radiomarquée vient s’hybrider sur le fragment ADN à séquencer. Des désoxyribonucléotides (dNTPs) pour les quatre bases sont ajoutés au mélange réactionnel ainsi qu’une Polymérase. Le mélange est divisé en quatre (un par base) et on rajoute chaque fois le didésoxyribonucléotide correspondant au mélange A, T, C et G. Chaque fois qu’un ddNTP se fixe pendant la réaction d’amplification, il bloque l’ajout d’une nouvelle base. Amorce. 1 ddNTP est statistiquement incorporé à toutes les positions possibles.")
5
Rappel: Méthode de Sanger
-Préparation du gel du polyacrylamide -Dénaturation et dépôt sur gel -Migration par électrophorèse -Révélation (S35 ou nitrate d’argent) ddG ddA ddT ddC Longueur typique de lecture : 400 nucléotides Durée totale de l’expérience : environ 3 jours On obtient des fragments de différentes longueurs qui sont mis à migrer sur gel haute résolution. On peut ainsi lire en partant du bas du gel vers le haut la séquence.
 ddG. ddA. ddT. ddC. Longueur typique de lecture : 400 nucléotides Durée totale de l’expérience : environ 3 jours. On obtient des fragments de différentes longueurs qui sont mis à migrer sur gel haute résolution. On peut ainsi lire en partant du bas du gel vers le haut la séquence.")
6
Un nouveau mode de préparation des échantillons et leur dépôt sur support miniaturisé
Amplification simultanée d’un grand nombre de fragments d’ADN Sur une bille (Roche) Le développement de technologies à haut débit repose sur la possibilité de préparer rapidement un grand nombre d’échantillons et les déposer en masse sur un support miniaturisé (array). Les deux techniques 454 et Solexa par exemple commencent par l’amplification simultanée d’un grand nombre de fragments d’ADN, générés par une ou des enzymes de restriction à partir de l’ADN de départ. Cette amplification était rendue nécessaire par la difficulté de générer un signal de détection luminescent suffisamment intense à partir d’une seule molécule. Les fragments générés sont liés à des adaptateurs, petits oligonucléotides de séquence connue, qui permettent leur amplification simultanément à partir d’amorces universelles. Le type d’amplification varie selon les méthodes. Dans le cas de la technologie 454, l’amplification se fait en émulsion en présence de micro-billes sur laquelle des amorces similaires aux adaptateurs ont été déposées. Statistiquement un seul fragment d’ADN se trouve en présence d’une bille dans chaque gouttelette d’émulsion, ce qui assure la spécificité d’amplification en surface des billes. Des millions de billes sont ensuite déposées sur une plaque de « picotitration ». Il en est de même dans la technique SOLiD. Dans le cas de la technologie Solexa, des amorces sont déjà fixées de façon aléatoire sur un support solide. Elles vont permettre l’amplification des d’ADN directement sur ce support solide, après attache des fragments d’ADN et liaison sur une amorce complémentaire à l’autre extrémité dans une configuration en « pont ». Tout fragment d’ADN se trouve ainsi amplifié directement sur le support. La dilution initiale assure une séparation claire de millions de spots d’ADN amplifiés sur ce support (appelé « flowcell »). Sur une lame de verre (Illumina)
 Le développement de technologies à haut débit repose sur la possibilité de préparer rapidement un grand nombre d’échantillons et les déposer en masse sur un support miniaturisé (array). Les deux techniques 454 et Solexa par exemple commencent par l’amplification simultanée d’un grand nombre de fragments d’ADN, générés par une ou des enzymes de restriction à partir de l’ADN de départ. Cette amplification était rendue nécessaire par la difficulté de générer un signal de détection luminescent suffisamment intense à partir d’une seule molécule. Les fragments générés sont liés à des adaptateurs, petits oligonucléotides de séquence connue, qui permettent leur amplification simultanément à partir d’amorces universelles. Le type d’amplification varie selon les méthodes. Dans le cas de la technologie 454, l’amplification se fait en émulsion en présence de micro-billes sur laquelle des amorces similaires aux adaptateurs ont été déposées. Statistiquement un seul fragment d’ADN se trouve en présence d’une bille dans chaque gouttelette d’émulsion, ce qui assure la spécificité d’amplification en surface des billes. Des millions de billes sont ensuite déposées sur une plaque de « picotitration ». Il en est de même dans la technique SOLiD. Dans le cas de la technologie Solexa, des amorces sont déjà fixées de façon aléatoire sur un support solide. Elles vont permettre l’amplification des d’ADN directement sur ce support solide, après attache des fragments d’ADN et liaison sur une amorce complémentaire à l’autre extrémité dans une configuration en « pont ». Tout fragment d’ADN se trouve ainsi amplifié directement sur le support. La dilution initiale assure une séparation claire de millions de spots d’ADN amplifiés sur ce support (appelé « flowcell »). Sur une lame de verre. (Illumina)")
7
Pyroséquençage (454 - Roche)
Préparation de la librairie : PCR d’ADN fixé sur bille en émulsion (1 fragment par bille) Dépôt des billes dans puits (1 bille/puit) Pyroséquençage : ajout d’une seule base, s’il y a incorporation alors il y a libération d’un phosphate qui va agir avec protéine qui émettra de la lumière Lavage puis ajout d’une deuxième base, etc. Cette diapositive illustre la technologie utilisée par le 454 GS-FLX (Roche). L’ADN est fragmenté et des adaptateurs sont ligués sur chaque fragment. On sélectionne ensuite les fragments ayant deux adaptateurs différents qui viendront s’hybrider sur une bille de quelques microns de diamètre. Une amplification est réalisée sur la bille dans une émulsion où chaque bulle contient une seule bille et le milieu réactif de l’amplification. Ces billes sont ensuite déposées sur une plaque où le diamètre des puits ne permet d’accepter qu’une seule bille. Des billes plus fines contenant sulfurylase et luciferase sont rajoutées et viennent fixer la plus grosse. La réaction de pyroséquençage peut alors commencer. On commence par ajouter une base (par exemple A) dans un liquide réactionnel qui passe sur la plaque. S’il y a incorporation de la base alors la réaction permet la libération d’un phosphate inorganique qui va réagir dans une réaction en chaîne avec la luciférase qui émettra alors un rayonnement capturé par la caméra CCD. Une fois la prise de vue réalisée, on inactive l’émission de lumière, on élimine le liquide réactionnel avec le reste de A non incorporé et on ajoute la seconde base (par exemple G). L’analyse des images à chaque prise de vue pour une position de la plaque permet d’identifier la séquence. En cas de répétition d’une base, le signal lumineux est plus intense et proportionnel au nombre de bases incorporées, dans la limite de 6 à 7 nucléotides identiques. C’est une des principales limitations de la technique. Metzker 2010
 Dépôt des billes dans puits (1 bille/puit) Pyroséquençage : ajout d’une seule base, s’il y a incorporation alors il y a libération d’un phosphate qui va agir avec protéine qui émettra de la lumière. Lavage puis ajout d’une deuxième base, etc. Cette diapositive illustre la technologie utilisée par le 454 GS-FLX (Roche). L’ADN est fragmenté et des adaptateurs sont ligués sur chaque fragment. On sélectionne ensuite les fragments ayant deux adaptateurs différents qui viendront s’hybrider sur une bille de quelques microns de diamètre. Une amplification est réalisée sur la bille dans une émulsion où chaque bulle contient une seule bille et le milieu réactif de l’amplification. Ces billes sont ensuite déposées sur une plaque où le diamètre des puits ne permet d’accepter qu’une seule bille. Des billes plus fines contenant sulfurylase et luciferase sont rajoutées et viennent fixer la plus grosse. La réaction de pyroséquençage peut alors commencer. On commence par ajouter une base (par exemple A) dans un liquide réactionnel qui passe sur la plaque. S’il y a incorporation de la base alors la réaction permet la libération d’un phosphate inorganique qui va réagir dans une réaction en chaîne avec la luciférase qui émettra alors un rayonnement capturé par la caméra CCD. Une fois la prise de vue réalisée, on inactive l’émission de lumière, on élimine le liquide réactionnel avec le reste de A non incorporé et on ajoute la seconde base (par exemple G). L’analyse des images à chaque prise de vue pour une position de la plaque permet d’identifier la séquence. En cas de répétition d’une base, le signal lumineux est plus intense et proportionnel au nombre de bases incorporées, dans la limite de 6 à 7 nucléotides identiques. C’est une des principales limitations de la technique. Metzker")
8
Séquençage en temps réel Utilisation de terminateurs réversibles (Solexa/ Illumina)
Dans le cas du du Genome Analyser de Solexa, l’ADN est fragmenté et ligué à des adaptateurs différents à chaque extrémité. Ces fragments s’hybrident sur une surface physique : la ‘flowcell’. Une amplification est réalisée et on se retrouve avec des colonies de fragment PCR identiques. La détection des nucléotides incorporés se fait alors base après base, quasiment en temps réel. On ajoute des amorces de séquençage, une polymérase et des dNTPs fluorescents bloqués en 3’ (de façon réversible). Le premier dNTPb se fixe sur chaque séquence et empêche le prolongement de l’élongation. La prise de vue par la caméra CCD permet d’identifier la première base pour chaque colonie de fragment PCR. Le dNTPb est transformé en dNTP et on refait un cycle d’élongation d’une seule base. L’analyse des images successives permet de redéfinir la séquence ADN du fragment. On réalise un séquençage cyclique des différentes colonies en parallèle avec une prise de vue à chaque cycle. On arrive donc avec de nombreux fichiers images (7000 à Go de mémoire) qui vont être analysés afin d’obtenir l’information de séquence (100 à 300 Go) Extrémité 3’ protégée - Ajoût d’un seul dNTP, identifié par fluorescence Déprotection et élimination de la fluorescence avant nouvel ajoût de dNTPS Tan 2006, Metzker2010
. Le premier dNTPb se fixe sur chaque séquence et empêche le prolongement de l’élongation. La prise de vue par la caméra CCD permet d’identifier la première base pour chaque colonie de fragment PCR. Le dNTPb est transformé en dNTP et on refait un cycle d’élongation d’une seule base. L’analyse des images successives permet de redéfinir la séquence ADN du fragment. On réalise un séquençage cyclique des différentes colonies en parallèle avec une prise de vue à chaque cycle. On arrive donc avec de nombreux fichiers images (7000 à Go de mémoire) qui vont être analysés afin d’obtenir l’information de séquence (100 à 300 Go) Extrémité 3’ protégée - Ajoût d’un seul dNTP, identifié par fluorescence. Déprotection et élimination de la fluorescence avant nouvel ajoût de dNTPS. Tan 2006, Metzker2010.")
9
Un nouveau mode de préparation des échantillons et leur dépôt sur support miniaturisé
Immobilisation d’un brin unique sans amplification: Hélicos Biosciences Pacific Biosciences D’autres techniques beaucoup plus récentes proposent la capture de millions de fragments d’ADN individuels, sans qu’il soit nécessaire de les amplifier avant la réaction de séquençage. Helicos Biosciences propose de fixer les fragments d’ADN individuels soit à partir d’amorces déposées sur un support solide, soit directement sur ce support (figures c et d) Pacific Biosciences pour sa part propose la capture de fragments d’ADN de grande taille directement par des ADN polymérases fixées sur le support constitué de détecteurs ZMW (zero-mode waveguide detectors) réduisant ainsi le volume d’observation (figure d). L’absence d’amplification réduit les mutations qui peuvent se produire au moment de l’amplification et les biais d’amplification sur les fragments riches en AT ou en GC. Elles requièrent également moins de matériel de départ.
 Pacific Biosciences pour sa part propose la capture de fragments d’ADN de grande taille directement par des ADN polymérases fixées sur le support constitué de détecteurs ZMW (zero-mode waveguide detectors) réduisant ainsi le volume d’observation (figure d). L’absence d’amplification réduit les mutations qui peuvent se produire au moment de l’amplification et les biais d’amplification sur les fragments riches en AT ou en GC. Elles requièrent également moins de matériel de départ.")
10
Détection de la fluorescence portée par un dNTP:
SMRT Technology, Pacific Biosciences Mesure en temps réel de l’activité de la polymérase - Single Molecule Real Time SMRTTM DNA Sequencing Technology (Pacific Biosciences) : Treffer 2010 La technologie SMRT (single molecule, real time) de Pacific Biosciences utilise une polymérase fixée au fond d’un puits d’une dizaine de nanomètres de diamètre et d’un volume de 20 zeptolitres (10-21 litres). Elle permet de séquencer un fragment simple brin en temps réel car une caméra ultra-précise se focalise sur la polymérase et enregistre chaque dNTP incorporé (marqué sur le phosphore). La puissance de cette technique est liée à la précision de la caméra à focaliser sur la polymérase et à la possibilité de faire abstraction du bruit de fond. Dans ce volume, l’activité d’un seul nucléotide peut être détecté parmi un bruit de fond de plusieurs centaines de nucléotides fluorescents. La vitesse de séquençage est de 10 bases / secondes et 1000 puits par puce (36Mb / heure), pour des lectures > 1000pb. Polymerisation avec nucléotides marqués sur P /fluorophore Zero-mode waveguides (détection fluo d’une seule mol.)
 : Treffer La technologie SMRT (single molecule, real time) de Pacific Biosciences utilise une polymérase fixée au fond d’un puits d’une dizaine de nanomètres de diamètre et d’un volume de 20 zeptolitres (10-21 litres). Elle permet de séquencer un fragment simple brin en temps réel car une caméra ultra-précise se focalise sur la polymérase et enregistre chaque dNTP incorporé (marqué sur le phosphore). La puissance de cette technique est liée à la précision de la caméra à focaliser sur la polymérase et à la possibilité de faire abstraction du bruit de fond. Dans ce volume, l’activité d’un seul nucléotide peut être détecté parmi un bruit de fond de plusieurs centaines de nucléotides fluorescents. La vitesse de séquençage est de 10 bases / secondes et 1000 puits par puce (36Mb / heure), pour des lectures > 1000pb. Polymerisation avec nucléotides marqués sur P /fluorophore. Zero-mode waveguides (détection fluo d’une seule mol.)")
11
Les rendements de séquençage évoluent très vite…
La longueur des fragments séquencés reste un gros problème des nouvelles technologies de séquençage. Cependant d’année en année ces longueurs augmentent de même que le débit de séquençage. HiSeq2000 avec kit TrueSeqv3-v4, 100 a 150 bases pair-end, 200 millions de cluster par lane (2x200 de reads en pair-end), 7 lanes en general (la 8e en control, soit 400 M x 150 x 7/run = 420 Gbases... (60/lanes) HiSeq2000 avec kit TrueSeqv3-v4, 100 a 150 bases pair-end, 200 millions de cluster par lane (2x200 de reads en pair-end), 7 lanes en general (la 8e en control, soit 400 M x 150 x 7/run = 420 Gbases... (60/lanes)
, 7 lanes en general (la 8e en control, soit 400 M x 150 x 7/run = 420 Gbases... (60/lanes) HiSeq2000 avec kit TrueSeqv3-v4, 100 a 150 bases pair-end, 200 millions de cluster par lane (2x200 de reads en pair-end), 7 lanes en general (la 8e en control, soit 400 M x 150 x 7/run = 420 Gbases... (60/lanes)")
12
Comparaison de quelques méthodes
Sanger 454/ Roche GS FLX Titanium Illumina/ Solexa Genetic Analyser Life:APG SOLiD SMRT/ Pacific Biosciences Clonage Oui Non Technique ddNTPs Pyro-séquençage Terminateurs réversibles Ligation Polymérisation en direct Molécule unique Lecture 800 bases/lecture 384 lectures/run 270 kb/run/3j 350 à 600 bases/lecture lectures/run 0.45 Gb /run/8h 75 ou 100 bases/lecture pls M lectures/run 14 Gb/run/ 4j 75 bases/lecture 100 à 200 M lectures/run 30 Gb/run/ 7j 1000 à 3000 bases/lecture et plus à venir 75000 lectures/run XX Gb / run / 30 min, Avantages Fiabilité et taille des fragments Taille des fragments (seq rep.) et rapidité Plateforme la plus utilisée Rapidité Encodage/ 2 bases limite erreurs Grand potentiel pour de gros fragments Incon-vénients Temps et coût Coût réactifs Erreurs / homo-polymères Indels Longueur des lectures Un peu plus long Grand taux d’erreurs Le tableau présente différentes propriétés des technologies ‘Next-Generation’ en comparaison avec la technologie Sanger. La quantité de données à gérer à la fin d’un run de séquençage ainsi que la taille des fragments pose cependant de nombreux problèmes au niveau du traitement bio-informatique de ces données. Par exemple l’algorithme BLAST qui permet d’aligner deux ou plusieurs séquences ne peut pas fonctionner avec des fragment de 26 bp car les scores qu’il atteint sont insuffisants au niveau statistique pour accepter un alignement. L’ensemble de ces techniques sont utilisables pour du reséquençage, ou du séquençage de novo. Le principal problème de la taille des séquences générées (13 bp à 250 bp, 450 pb pour la technique de Sanger), et donc de la taille des données générées. Un logiciel d’assemblage performant est nécessaire si l’on veut pouvoir contiguer les séquences produites et inférer des séquences de plus grande longueur. Finalement, certaines techniques sont complémentaires et parfois utilisées en combinaison pour des séquençages de génomes complets: 454 + Solexa 454 + SOLiD
 et rapidité. Plateforme la plus utilisée. Rapidité. Encodage/ 2 bases limite erreurs. Grand potentiel pour de gros fragments. Incon-vénients. Temps et coût. Coût réactifs. Erreurs / homo-polymères. Indels. Longueur des lectures. Un peu plus long. Grand taux d’erreurs. Le tableau présente différentes propriétés des technologies ‘Next-Generation’ en comparaison avec la technologie Sanger. La quantité de données à gérer à la fin d’un run de séquençage ainsi que la taille des fragments pose cependant de nombreux problèmes au niveau du traitement bio-informatique de ces données. Par exemple l’algorithme BLAST qui permet d’aligner deux ou plusieurs séquences ne peut pas fonctionner avec des fragment de 26 bp car les scores qu’il atteint sont insuffisants au niveau statistique pour accepter un alignement. L’ensemble de ces techniques sont utilisables pour du reséquençage, ou du séquençage de novo. Le principal problème de la taille des séquences générées (13 bp à 250 bp, 450 pb pour la technique de Sanger), et donc de la taille des données générées. Un logiciel d’assemblage performant est nécessaire si l’on veut pouvoir contiguer les séquences produites et inférer des séquences de plus grande longueur. Finalement, certaines techniques sont complémentaires et parfois utilisées en combinaison pour des séquençages de génomes complets: Solexa SOLiD.")
13
Ce qui change avec les NGS…
Une grande quantité de données en un temps record (un génome par individu possible) Ce qui change avec les NGS, c’est la grande quantité de données qu’il est possible de générer en peu de temps. Le séquençage de novo d’un grand nombre d’espèces végétales, pourtant parmi les plus gros existant, semble maintenant « théoriquement » réalisable, si l’on fait abstraction des problèmes de séquences répétées et de polyploidie…
 Ce qui change avec les NGS, c’est la grande quantité de données qu’il est possible de générer en peu de temps. Le séquençage de novo d’un grand nombre d’espèces végétales, pourtant parmi les plus gros existant, semble maintenant « théoriquement » réalisable, si l’on fait abstraction des problèmes de séquences répétées et de polyploidie…")
14
Ce qui change avec les NGS…
Une grande quantité de données en un temps record (un génome par individu possible) Des séquences courtes (de + en + longues) mais une grande redondance (voir les différences et les distinguer des erreurs) Le coût (de moins en moins cher) Le paradigme de stockage - séquençage
 Des séquences courtes (de + en + longues) mais une grande redondance (voir les différences et les distinguer des erreurs) Le coût (de moins en moins cher) Le paradigme de stockage - séquençage.")
15
Les grandes questions posées en amélioration des plantes
Peut on optimiser la caractérisation et l’exploitation de la diversité déjà disponible? Ressources génétiques Peut on créer de la variation génétique « utile » plus facilement? Recombinaison Peut on diminuer le temps de sélection/fixation ? Sélection/Fixation Comment multiplier rapidement et assainir les variétés obtenues? Variété Source : teosinte.wisc.edu

16
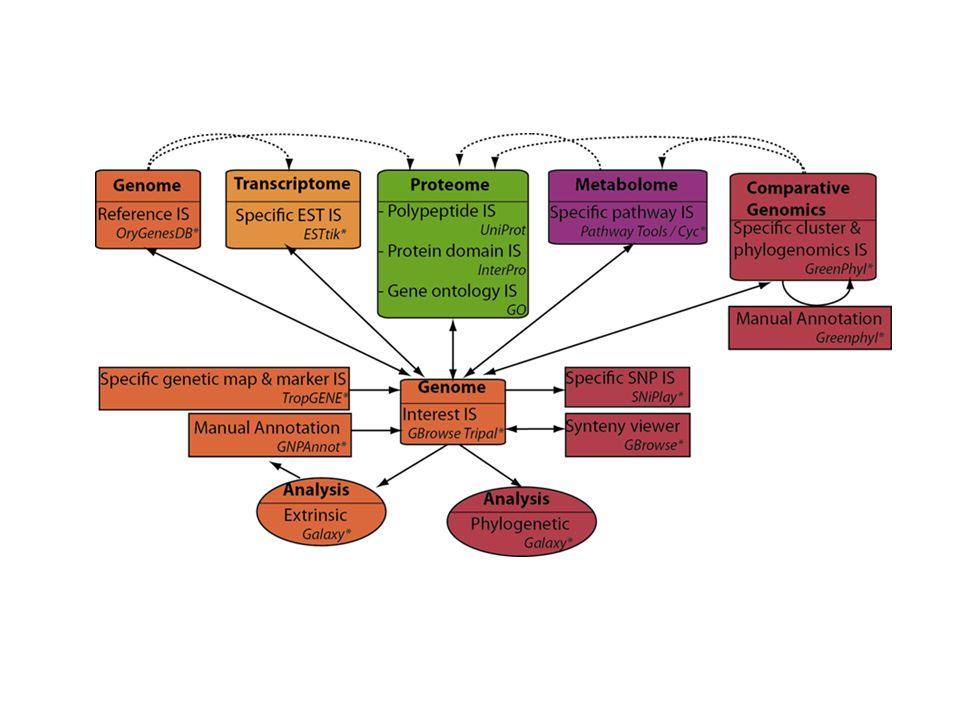
Des avancées méthodologiques et scientifiques valorisables
Biotechnologies cellulaires et moléculaires Ressources génétiques Génotypage Analyse des phénotypes Recombinaison Concepts en génétique et bio-mathématiques pour l’analyse de données complexes Sélection/Fixation Variété

17
Les NGS: Une innovation importante à intégrer dans ce schéma.
Caractériser la diversité disponible Marqueurs +++ Ressources génétiques Mieux comprendre le fonctionnement des plantes cultivées Séquençage / annotation, expression, épigénétique Recombinaison Identifier des polymorphismes d’intérêt agronomique Marqueurs / QTL et génétique d’association Sélection/Fixation Faciliter le suivi de nouvelles combinaisons alléliques Sélection assistée par marqueurs Mais de nouvelles problématiques (bioinformatiques) pour traiter ce flux de données sans précédent… Variété
 pour traiter ce flux de données sans précédent… Variété.")
18
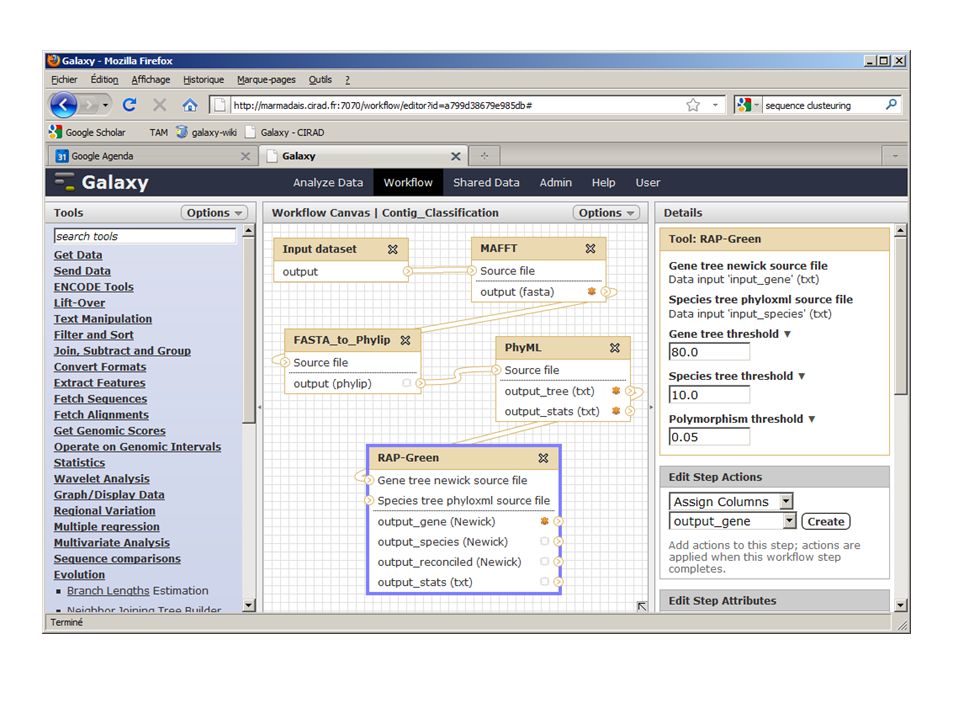
Objet de la formation: Analyse bioinformatique de séquences pour l’amélioration des plantes
Des données NGS nous arrivent en masse Projet ARCAD (transcriptomes) Projets de séquençage (de-novo) de génomes complets Projets de re-séquençage Il faut pouvoir les traiter convenablement… (étape limitante) Présentation De démarches d’analyse autour des séquences biologiques du système d’un système d’information (Galaxy) capable d’effectuer des chaines de traitement de façon simplifiée
 Projets de séquençage (de-novo) de génomes complets. Projets de re-séquençage. Il faut pouvoir les traiter convenablement… (étape limitante) Présentation. De démarches d’analyse autour des séquences biologiques. du système d’un système d’information (Galaxy) capable d’effectuer des chaines de traitement de façon simplifiée.")
19
Next-generation sequencing
Analyses NGS Next-generation sequencing Aujourd’hui, dans nos labos, les bioinformaticiens peuvent faire des analyses complètes de type Transcriptomiques (ArCad) Reséquençages génomiques (GrapeReseq) Demain : Séquençage de nouveaux génomes Métagénomique (écosystèmes) Après-demain: Séquençage cellule / cellule … Concernant les analyses de séquençage de nouvelle génération, aujourd’hui, dans nos labos, les bioinformaticiens sont capables de faire des analyses complète De Transcriptomes c’est le cas du projet Arcad et de reséquençage c’est du projet GrapeReseq sur la vigne C’est aussi le cas du riz ARCAD Grégory CARRIER, Loïc Le Cunff UMT Géno-Vigne, IFV-INRA-Montpellier SupAgro “Identification of the molecular polymorphism responsible for the clonal variation in grapevine” ANR GrapeReseq en cours Vitinext (500 ANR Biotechno) Riz ARCAD glaberima facteur 10 en perspective génomes Sequençage de nouveau génomes champignons magnaporteae
 Reséquençages génomiques (GrapeReseq) Demain : Séquençage de nouveaux génomes. Métagénomique (écosystèmes) Après-demain: Séquençage cellule / cellule. … Concernant les analyses de séquençage de nouvelle génération, aujourd’hui, dans nos labos, les bioinformaticiens sont capables de faire des analyses complète. De Transcriptomes c’est le cas du projet Arcad et de. reséquençage c’est du projet GrapeReseq sur la vigne. C’est aussi le cas du riz ARCAD. Grégory CARRIER, Loïc Le Cunff UMT Géno-Vigne, IFV-INRA-Montpellier SupAgro Identification of the molecular polymorphism responsible for the clonal variation in grapevine ANR GrapeReseq en cours Vitinext (500 ANR Biotechno) Riz ARCAD glaberima facteur 10 en perspective génomes. Sequençage de nouveau génomes champignons magnaporteae.")
21
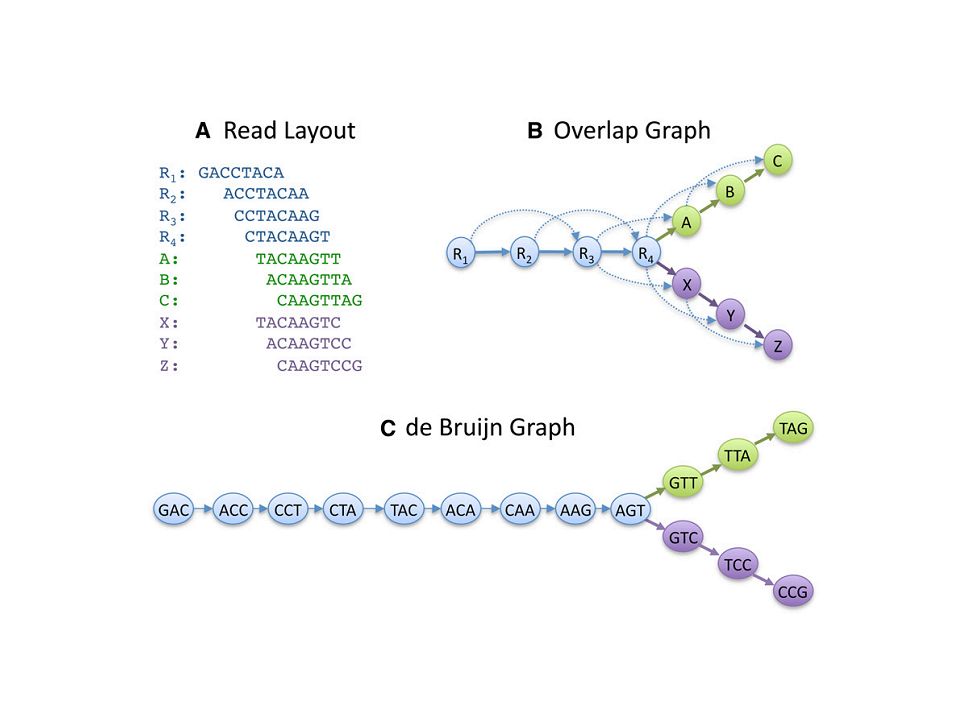
de novo assembling

23
How to apply de Bruijn graphs to genome assembly, Phillip E C Compeau ,Pavel A Pevzner , Glenn Tesler Nature Biotechnology, 29,, 987–991 (2011)
")
24
Mapping Maq Bowtie Trapnell C, Salzberg SL: How to map billions of short reads onto genomes. Nat Biotechnol 2009, 27(5):
:")
29
Natural selection vs user selection of the best tools

30
The rate of increase… Manipulation des fichiers, stockage : problème Coût en disques Besoin en Calculs Format des données Stein LD: The case for cloud computing in genome informatics. Genome Biol 2010, 11(5):207.
:207.")
31
Stein, L.D. (2008) Towards a cyberinfrastructure for the biological sciences: progress, visions and challenges, Nat Rev Genet,
 Towards a cyberinfrastructure for the biological sciences: progress, visions and challenges, Nat Rev Genet,.")
32
cyberinfrastructure will be an absolutely indispensable
“ in a decade the cyberinfrastructure will be an absolutely indispensable part of the biological researcher’s equipment” Stein, L.D. (2008) Towards a cyberinfrastructure for the biological sciences: progress, visions and challenges, Nat Rev Genet, “biological researchers will need to become familiar with the basics of computer science,[…], and have the skills to put this information in a form that can be readily adapted and re‑used by others in the community. This will require changes in the way biology is taught at the undergraduate and graduate levels”
 Towards a cyberinfrastructure for the biological sciences: progress, visions and challenges, Nat Rev Genet, biological researchers will need to become familiar with the basics of computer science,[…], and have the skills to put this information in a form that can be readily adapted and re‑used by others in the community. This will require changes in the way biology is taught at the undergraduate and graduate levels")
33
cyberinfrastructure will be an absolutely indispensable
“ in a decade the cyberinfrastructure will be an absolutely indispensable part of the biological researcher’s equipment” Stein, L.D. (2008) Towards a cyberinfrastructure for the biological sciences: progress, visions and challenges, Nat Rev Genet, “biological researchers will need to become familiar with the basics of computer science,[…], and have the skills to put this information in a form that can be readily adapted and re‑used by others in the community. This will require changes in the way biology is taught at the undergraduate and graduate levels”
 Towards a cyberinfrastructure for the biological sciences: progress, visions and challenges, Nat Rev Genet, biological researchers will need to become familiar with the basics of computer science,[…], and have the skills to put this information in a form that can be readily adapted and re‑used by others in the community. This will require changes in the way biology is taught at the undergraduate and graduate levels")
34
Part of this global cyberinfrastructure

35
Ressources de calculs et de stockage
La plateforme repose en partie sur un cluster de calculs pouvant héberger 65 To de données, avec 240 processeurs, machines de 32 à 96 Go de RAM.

36
Funded by the French National Research Agency ANR (2008-2010)
Stéphanie Sidibe Bocs Funded by the French National Research Agency ANR ( ) CIRAD, Bioversity & INRA Community annotation system (CAS) of structural and functional annotation Automatic predictions and manual curations of genes and transposable elements Based on GMOD components 36
 CIRAD, Bioversity & INRA. Community annotation system (CAS) of structural and functional annotation. Automatic predictions and manual curations of genes and transposable elements. Based on GMOD components. 36.")
37
International Nucleotide Sequence Database Collaboration
The International Nucleotide Sequence Databases (INSD) have been developed and maintained collaboratively between DDBJ, ENA, and GenBank for over 18 years. The INSDC advisory board, the International Advisory Committee , is made up of members of each of the databases' advisory bodies. At their most recent meeting, members of this committee unanimously endorsed and reaffirmed the existing data-sharing policy of the three databases that make up the INSDC, which is stated below. 37
 have been developed and maintained collaboratively between DDBJ, ENA, and GenBank for over 18 years. The INSDC advisory board, the International Advisory Committee , is made up of members of each of the databases advisory bodies. At their most recent meeting, members of this committee unanimously endorsed and reaffirmed the existing data-sharing policy of the three databases that make up the INSDC, which is stated below. 37.")
38
automated procedures (auto-set qualifiers, transposable element structure)
validation procedures (structure, sequence content & length, introns, qualifiers and mandatory fields) generalisation of Controlled Vocabulary 38
 generalisation of Controlled Vocabulary. 38.")
39
Gaëtan Droc

40
Jean-François Dufayard
16 plant species gene families genes v2.0 Matthieu Conte Jean-François Dufayard “I suggest that functional predictions can be greatly improved by focusing on how the genes became similar in sequence (i.e., evolution) rather than on the sequence similarity itself”. Jonathan A. Eisen 1998 GreenPhylDB use phylogenomic method to identify homologous genes A phylogenomic system for plant comparative genomics GreenPhylDB v2.0 A comparative genomic database for crop functional genomics Statistics from 2010_02_GreenPhyl_Colombie_MRuiz.ppt Mathieu Rouard 40 40
 rather than on the sequence similarity itself . Jonathan A. Eisen GreenPhylDB. use phylogenomic method to identify homologous genes. A phylogenomic system for plant comparative genomics. GreenPhylDB v2.0. A comparative genomic database for crop functional genomics. Statistics from 2010_02_GreenPhyl_Colombie_MRuiz.ppt. Mathieu Rouard")
41
Xavier Argout Functional annotation Marilyne Summo

42
SNIPlay A web-based tool for SNP and polymorphism analysis. From sequencing traces, alignment or allelic data given as input, it detects SNP and insertion/deletion events, and sends sequences and allelic data to an integrative pipeline (haplotype reconstruction, haplotype network, LD, diversity) Aussi phylogeny.fr Alexis Dereeper
 Aussi phylogeny.fr. Alexis Dereeper.")
46
Strategy : comparative population genomics with transcriptomics data
Available reference ? genome/transcriptome No Yes 454 sequencing Solexa sequencing Solexa sequencing De novo reference assembly Mapping on reference Ortholog/paralogs assignation Quite general strategy in population comparative genomics Polymorphism database in adapted format redundancy open reading frame CDS/UTR Diversity study Comparative domestication Life history trait impact Functionnal evolution CROP Breeding SNP database functional annotation selection footprint

47
Modélisation 3D par homologie
It is generally admitted that a reliable model can be obtain up from 25-30% sequence identity between template and target

48
Structures 3D des nsLTP 1MID (Type I) 1TUK (Type II) 2RKN (Type IV)
Le fold ne change pas fondamentalement 1MID (Type I) 1TUK (Type II) 2RKN (Type IV) Hordeum vulgare Triticum aestivum Arabidopsis thaliana C1-C6 C5-C8 C1-C5 C6-C8 C1-C5 C6-C8 48
 1TUK. (Type II) 2RKN. (Type IV) Hordeum vulgare. Triticum aestivum. Arabidopsis thaliana. C1-C6. C5-C8. C1-C5. C6-C8. C1-C5. C6-C")
49
Impala Méthode (Monte Carlo) de prédiction de capacité d’interaction avec une memb, validé par données de mutagénèse dirigée On peut avoir dimères+lipides+interaction mb 49

52
Thanks to Equipe Intégration Des Données, UMR AGAP

53
Thank you

Présentations similaires