Télécharger la présentation
1
OBSERVATION CLINIQUE

2
MOTIF D’HOSPITALISATION
Nouveau-né transféré à j1 de vie par MAT CHU pour anomalie des organes génitaux externes ACTD MATERNELS Mère née en 1973 G5P4(2 garçons, 1 fille et 1 avortement) Pas d’ATCD familiaux
 Pas d’ATCD familiaux.")
3
GROSSESSE ACTUELLE Suivie à Reims
Sérologies rubéole + toxo+VIH- syphilis- VHB VHC- ECBV : RAS A 7 mois, échographie réalisée : fémur court OGE douteux Amniocentèse : 45 XO probable syndrome de Turner 48h avant accouchement : F.I.S.H Y+

4
ACCOUCHEMENT Césarienne pour utérus bi cicatriciel
Accouchement sans particularité

5
ETAT DE L’ENFANT A LA NAISSANCE
Terme : 39 SA Poids : 3170g Taille : 48cm PC : 33cm Apgar : BAI

6
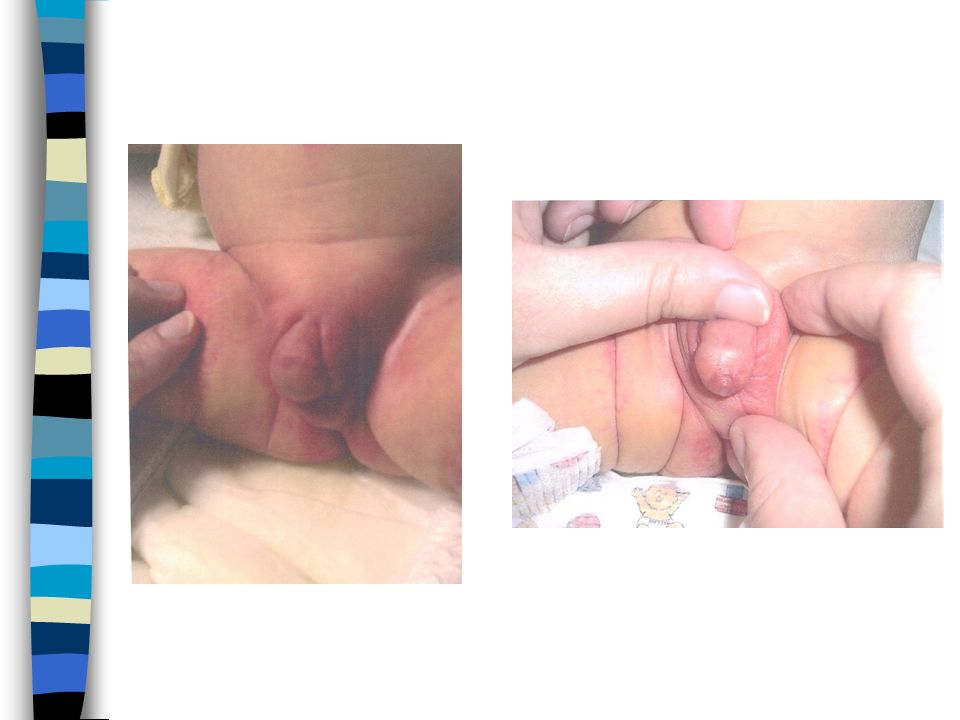
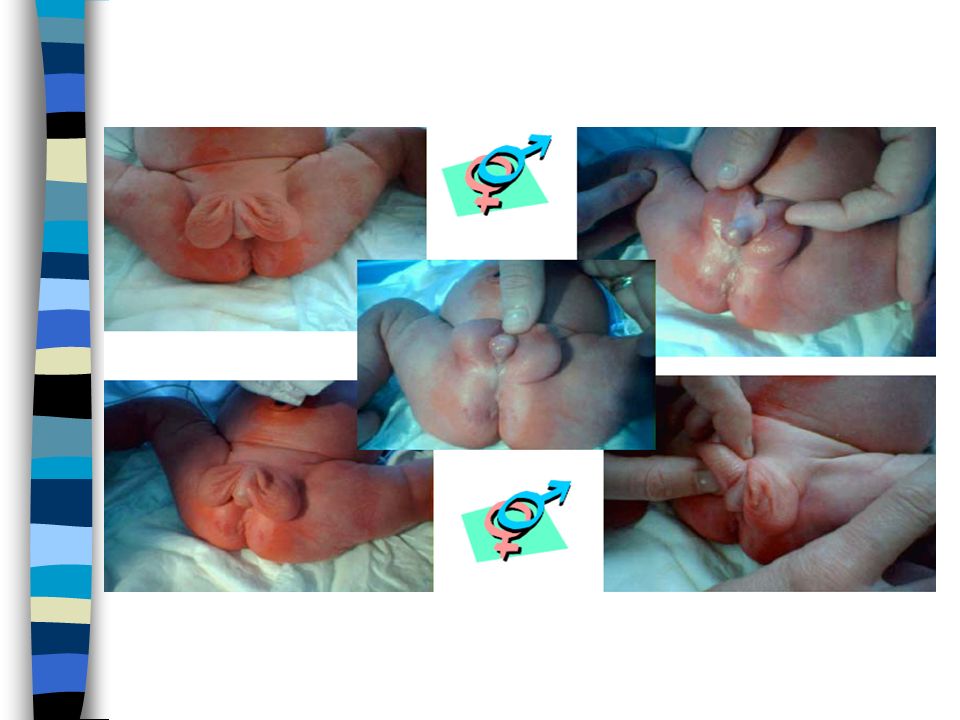
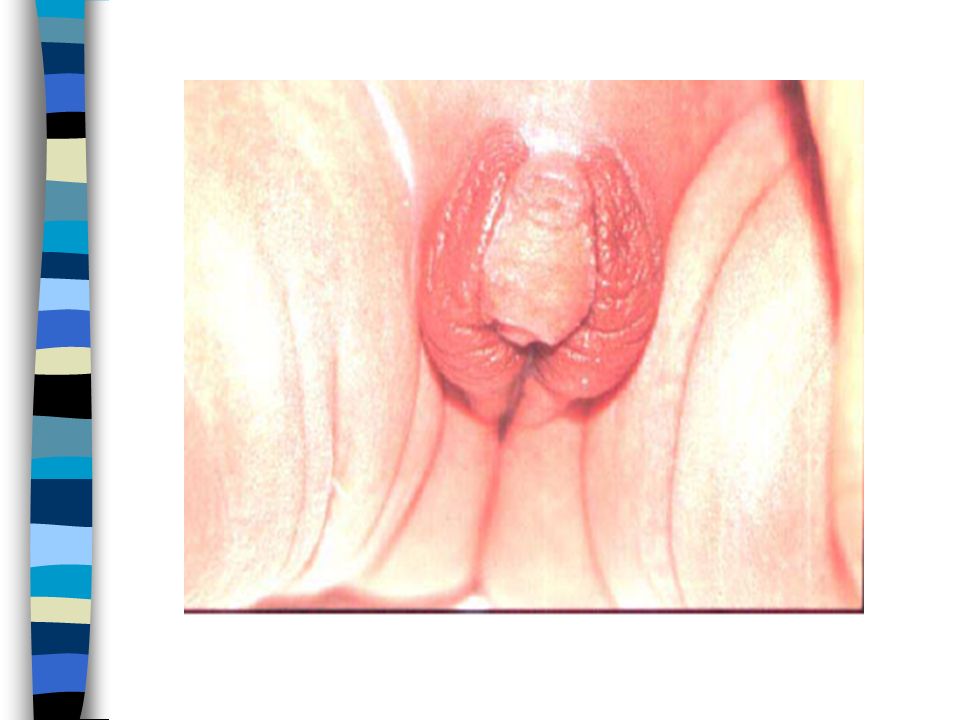
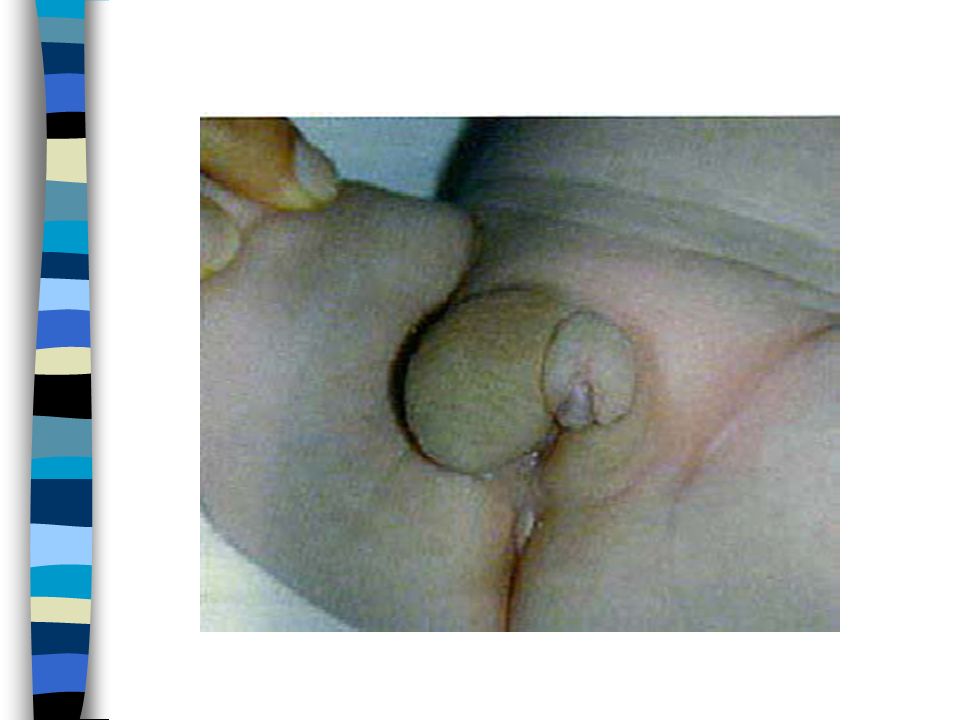
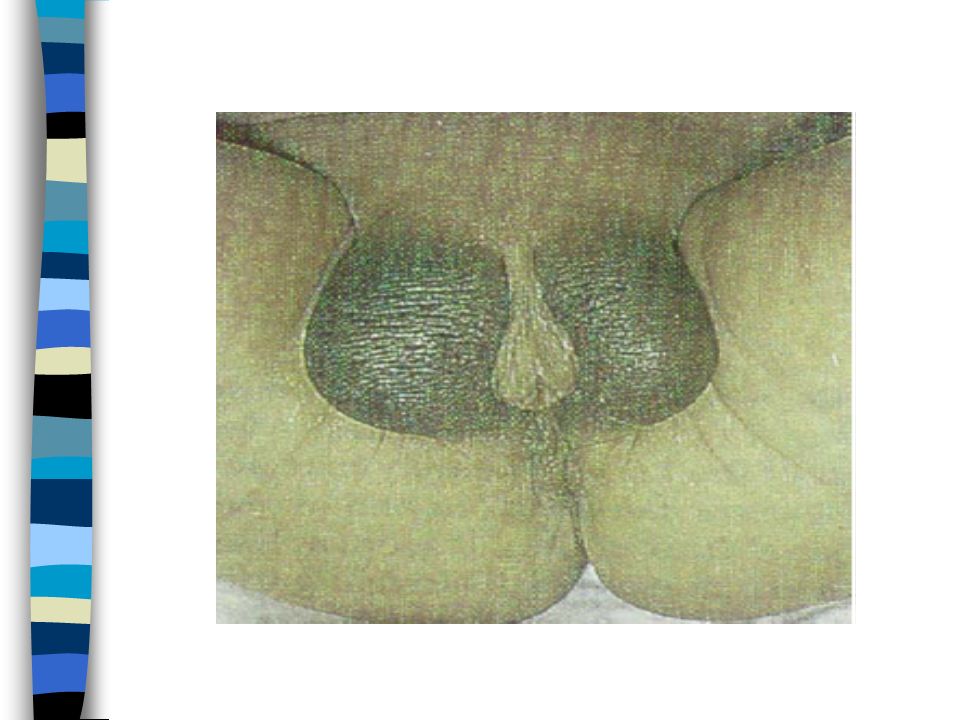
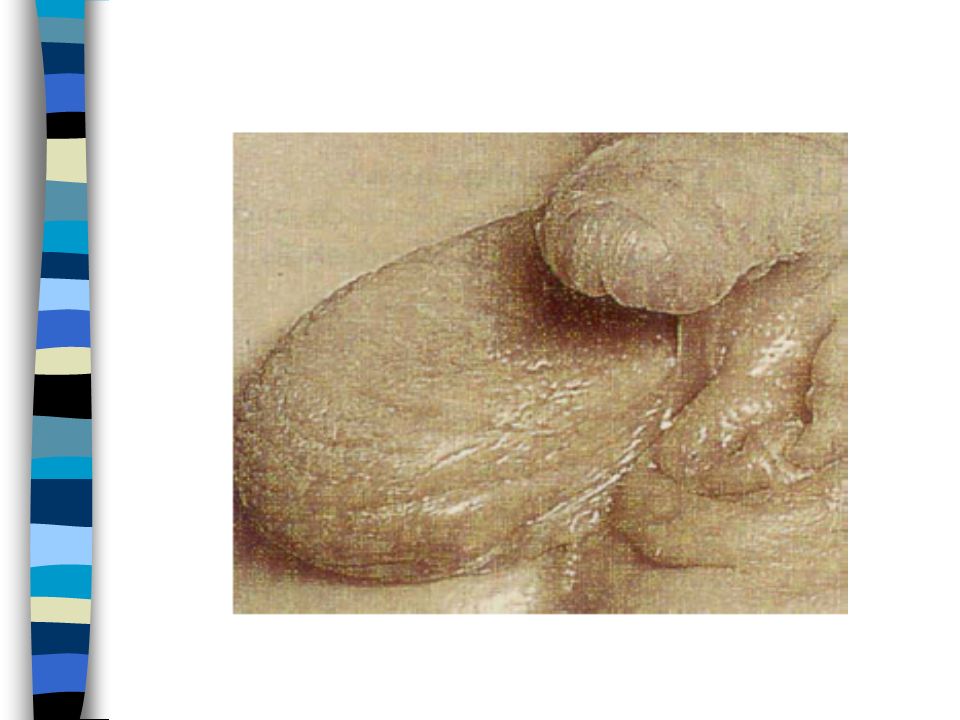
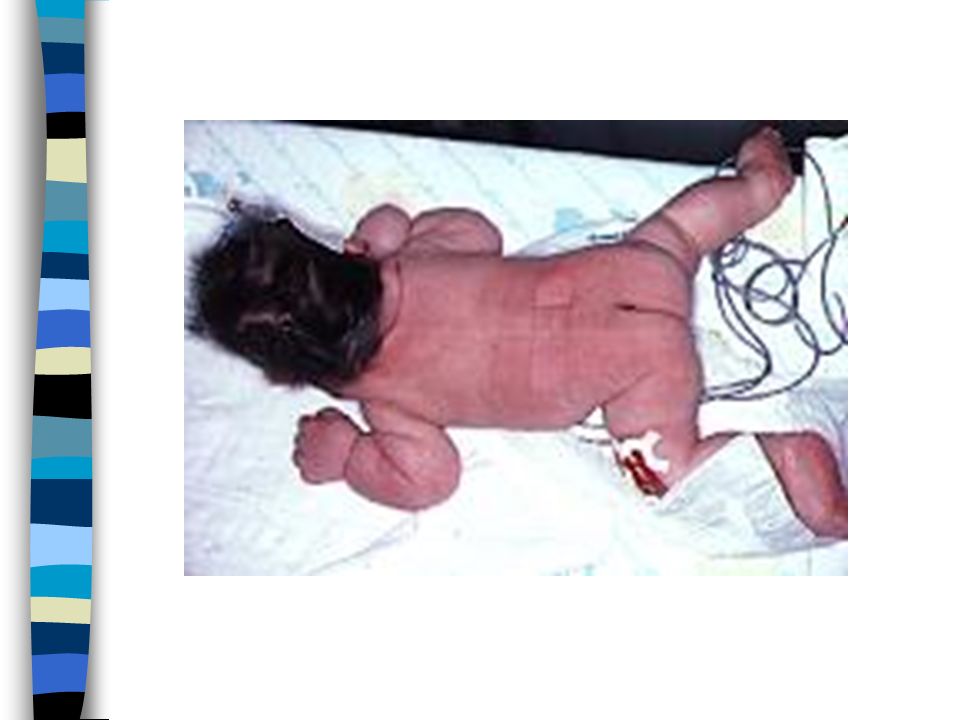
EXAMEN CLINIQUE => Ambiguïté sexuelle : Masculanisation stade 4 Prader Bourgeon génital pénien Ebauche de grandes lèvres avec fusion posterieure Hypospadias ventral Pas de gonades palpables Pas d’hyperpigmentation

8
BILAN REALISE Bilan bio : normal Bilan hormonal :
17OHP normale(<60nmol/l) Testostérone 2.1ng/ml(garçon<3ng/ml,fille indétectable) delta 4 androstène dione, FSH LH, SDHA normales Échographie abdomino-pelvienne : utérus antéfléchi antéversé urètre type masculin pas de gonades visualisées surrénales de grande taille
 Testostérone 2.1ng/ml(garçon<3ng/ml,fille indétectable) delta 4 androstène dione, FSH LH, SDHA normales. Échographie abdomino-pelvienne : utérus antéfléchi antéversé. urètre type masculin. pas de gonades visualisées. surrénales de grande taille.")
9
Génitographie et scopie :
Existence d’une cavité vaginale surmontée d’un col utérin en sous vésical immédiat Existence d’une probable cavité utérine Caryotype post natal : Mosaïque : 86% 45 XO 14% 46 XY

10
CONCLUSION Dysgénésie gonadique mixte équivalente de syndrome de Turner/mosaique Décision d’orientation vers sexe féminin PEC : plastie de féminisation avec abaissement urétro-vaginale vers 6 mois gonadectomie bilatérale à distance

11
CAT DEVANT UNE AMBIGUITE SEXUELLE

12
Introduction Physiopathologie Démarche diagnostique Étiologies Syndrome de TURNER Bibliographie

13
Le plus souvent diagnostic néonatal Urgence :
INTRODUCTION Le plus souvent diagnostic néonatal Urgence : Métabolique (syndrome de perte de sel) Attribuer sexe à l’enfant (délai légal de 3 jours)
 Attribuer sexe à l’enfant (délai légal de 3 jours)")
14
PHYSIOPATHOLOGIE Sexe masculin
Présence chez le fœtus du gène SRY sur chromosome Y qui détermine la gonade en testicule Testicule fœtal sécrète 2 hormones : AMH sécrétée par cellules de Sertoli provoquant régression de canaux de Muller Testostérone sécrétée par cellules de Leydig entraînant le dev des canaux de Wolff

15
Conduits génitaux internes dérivent des canaux de Wolff : différenciation en épididyme/canaux déférents/vésicules séminales/canaux éjaculateurs Canaux de Muller régressent : résidus d’hydatide

16
Sexe féminin Pas de chromosome Y portant le gène SRY
Canaux de Muller : Trompes puis utérus par fusion Canaux de Wolff régressent(utricule)
")
17
DEMARCHE DIAGNOSTIQUE
Interrogatoire ACTD FAMILIAUX Consanguinité ATCD d’ambiguïté sexuelle Déficits enzymatiques génétiques Prise de médicaments

18
Examen physique OGE Bourgeon génital +orifices uro-génitaux
Présence de gonades Aspect de la peau génitale : plis / pigmentation Description selon Classification de PRADER

19
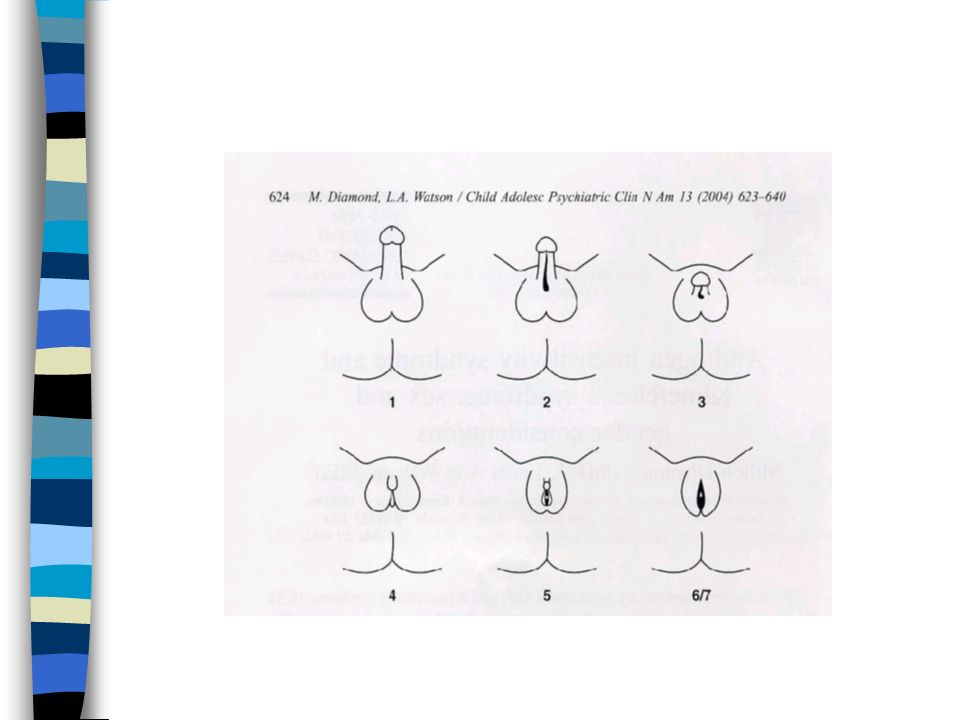
Classification de PRADER
1. Vulve normale avec hypertrophie clitoridienne 2. Large vestibule en entonnoir s’ouvrant sur à la base du clitoris. Gdes lèvres séparées ou partiellement soudées 3. Clitoris volumineux sur lequel s’ouvre un sinus uro génital étroit dans lequel se jettent uretre et vagin. Gdes lèvres partiellement soudées 4. Aspect de garçon avec verge hypoplasique et souvent coudée. Hypoplasie périnéale. Aspect d’hypospade : sinus urogénital dans lequel s’abouche un vagin hypoplasique. Gdes lèvres soudées 5. Aspect de garçon cryptorchide.Vagin s’abouche haut dans l’urètre

22
Signes associés Bilan biologique Dysmorphie faciale Hypospadias
Cryptorchidie Hypertrophie clitoridienne Examen clinique complet Bilan biologique Ionogramme sanguin et urinaire

23
Bilan hormonal minimum
17OH progestérone Testostérone Delta 4 androstène dione,composé S,SDHA FSH/LH ACTH,renine cortisol Caryotype haute résolution avec étude biologie moléculaire

24
Bilan morphologique Échographie abdomino-pelvienne :
Recherche d’un utérus ou de reliquat utérin, d’ovaires Étude des surrénales

25
Génitoscopie et génitographie :
Étude de la cavité vaginale Recherche d’empreinte du col utérin Morphologie et longueur de l’urètre Coelioscopie exploratrice : définir l’anatomie interne plus précisément

26
Pseudo-hermaphrodisme masculin (35%)
ETIOLOGIES Pseudo-hermaphrodisme masculin (35%) Gonade comportant tissu testiculaire Caryotype : 46XY Présence de 2 testicules Aspects variables des OGI et OGE
 Gonade comportant tissu testiculaire. Caryotype : 46XY. Présence de 2 testicules. Aspects variables des OGI et OGE.")
27
Défaut de développement du testicule (15%)
Défaut de détermination testiculaire Défaut de différenciation testiculaire : Testicules dysgénétiques bilatéraux Syndrome de régression testiculaire Dysgénésie gonadique

28
Insuffisance de production de la testostérone
Insuffisance d’action des hormones gonadotropes(agénésie des cellules de Leydig,déficit en LH,mutation des récepteurs LH) Anomalie du métabolisme de la testostérone(déficit en 5 alpha réductase)
 Anomalie du métabolisme de la testostérone(déficit en 5 alpha réductase)")
30
Résistance à la testostérone (60%)
Récepteurs défectueux Syndrôme d’insensibilité aux androgènes Syndrome de persistance des dérivés Mulleriens Mutation de l’AMH ou de son récepteur Idiopathiques (20%)
")
31
Pseudo-hermaphrodisme féminin (60%)
Gonade comportant tissu ovarien Caryotype=46XX Présence de 2 ovaires OGI plutôt féminins OGE variables

32
Origine fœtale Hyperplasie congénitale des surrénales
Dysgénésie gonadique Tumeurs surrénaliennes/CUSHING néonatal Déficits enzymatiques(aromatase,P450oxydo réductase)
")
36
Origine maternelle Tumeur ovarienne/surrénaliennes Cushing Lutéome gravidique Médicaments Idiopathiques

37
Hermaphrodisme vrai (5%)
=> Coexistence dans les gonades de tissu ovarien et testiculaire actif : Caryotype 46XX (60% des cas) Caryotype 46XY (10% des cas) Caryotype mosaïque (30% des cas)
 Caryotype 46XY (10% des cas) Caryotype mosaïque (30% des cas)")
39
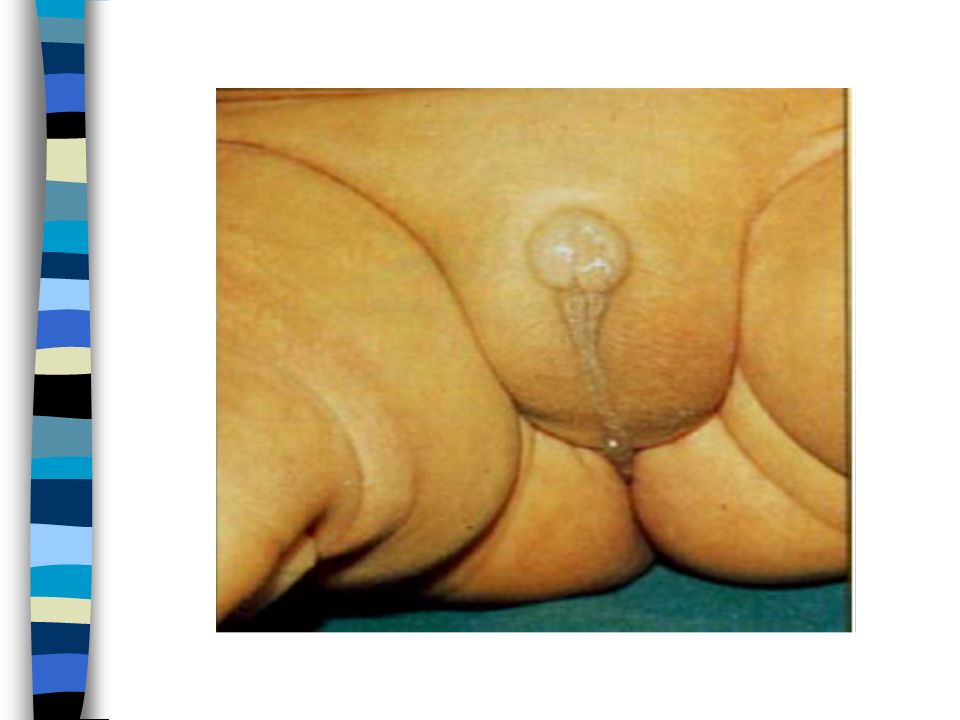
SYNDROME DE TURNER Clinique 1 femme sur 4000
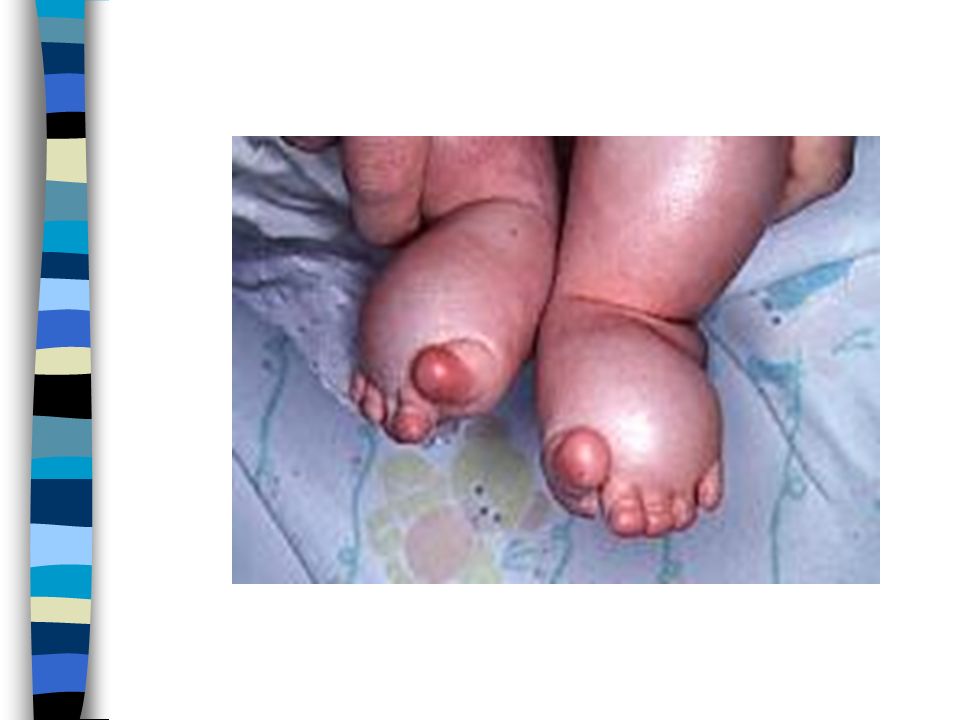
Avant naissance : Hygroma cervical ou oedèmes généralisés Naissance : Dysmorphie variable=visage triangulaire,cou court,fentes palpébrales obliques en bas et en dehors,ptosis,rétrognathisme,ptérygium coli) Oedème mains et pieds Diminution de la taille
 Oedème mains et pieds. Diminution de la taille.")
43
Rein : unique /en fer à cheval
Os : Déminéralisation osseuse Endocrinien : DID,obésité,hypothyroïdie C.V : atteinte aortique le plus souvent Otites à répétition,strabisme,dysplasie de hanche,scoliose Capacités intellectuelles normales

44
Diagnostic : Caryotype
55% XO 20% MOSAIQUE 25% XX (avec un des deux altéré)
")
45
Traitement Corriger insuffisances hormonales
Surveiller troubles métaboliques Chirurgie des malformations Ttnt par GH dès l’enfance (gain d’1.5 cm par an si débuté entre 7 et 12 ans) Ttnt par œstrogène puis œstrogène et progestérone pour dev pubertaire
 Ttnt par œstrogène puis œstrogène et progestérone pour dev pubertaire.")
46
BIBLIOGRAPHIE www.sfip-radiopédiatrie.org/EPOTIMOO/GEOTIMOO.htm
DIU endocrinologie(2007)
")