Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
J’ai fait séquencer mes petits ARN. Et Maintenant ?
Introduction à l’analyse des données de séquençage à haut débit en génomique fonctionnelle. 28 mars 2012, 15:30 – 17:00 J’ai fait séquencer mes petits ARN. Et Maintenant ?

2
Les trois principales classes de petits ARNs chez la drosophile
met Hen1 Produits des snoRNA, tRNA, rRNA. 2S Droso (30nt) +
 +")
3
small RNA deep sequencing
(Biases) 20-30nt RNA gel purification Library “Bar coding”
 20-30nt RNA gel purification. Library Bar coding")
4
Que Puis-je Faire avec mes séquences de petits ARN ?
Annotation Visualisation Découverte de loci Quantification d’expression Analyse structurale des précurseurs, signatures, … Mise en évidence de « visiteurs » (virus, …) … Informatique Bioinformatique
 … Informatique Bioinformatique.")
5
Matériel Un fichier de séquence au format fastq
Un ordinateur avec ~ 8 Mo RAM Un « Operating System Unix compliant » Un maniement confortable de cet OS Quelques logiciels génériques très utiles Un « vrai » éditeur de texte (TextWrangler, etc..) R, Gnuplot … Une bonne connaissance du web Le maniement niveau Débutant++ d’un langage de programmation Perl Python
 R, Gnuplot. … Une bonne connaissance du web. Le maniement niveau Débutant++ d’un langage de programmation. Perl. Python.")
6
Que contient le gros fichier fastq que j’ai téléchargé (et décompressé) ?
* Limite max pour ouvrir un gros fichier texte (~1.2 Go) Terminal Unix. Naviguer dans le dossier qui contient le fichier Taper la commande more <nom_du_fichier> lbcd-05:GKG13demo deepseq$ more GKG-13.fastq @HWIEAS210R_0028:2:1:3019:1114#AGAAGA/1 TNGGAACTTCATACCGTGCTCTCTGTAGGCACCATCAA +HWIEAS210R_0028:2:1:3019:1114#AGAAGA/1 bBb`bfffffhhhhhhhhhhhhhhhhhhhfhhhhhhgh @HWIEAS210R_0028:2:1:3925:1114#AGAAGA/1 TNCTTGGACTACATATGGTTGAGGGTTGTACTGTAGGC +HWIEAS210R_0028:2:1:3925:1114#AGAAGA/1 ]B]VWaaaaaagggfggggggcggggegdgfgeggbab @HWIEAS210R_0028:2:1:6220:1114#AGAAGA/1 +HWIEAS210R_0028:2:1:6220:1114#AGAAGA/1 aB^^afffffhhhhhhhhhhhhhhhhhhhhhhhchhhh @HWIEAS210R_0028:2:1:6252:1115#AGAAGA/1 +HWIEAS210R_0028:2:1:6252:1115#AGAAGA/1 aBa^\ddeeehhhhhhhhhhhhhhhhghhhhhhhefff @HWIEAS210R_0028:2:1:6534:1114#AGAAGA/1 TNAATGCACTATCTGGTACGACTGTAGGCACCATCAAT +HWIEAS210R_0028:2:1:6534:1114#AGAAGA/1 aB\^^eeeeegcggfffffffcfffgcgcfffffR^^] @HWIEAS210R_0028:2:1:8869:1114#AGAAGA/1 GNGGACTGAAGTGGAGCTGTAGGCACCATCAATAGATC +HWIEAS210R_0028:2:1:8869:1114#AGAAGA/1 aBaaaeeeeehhhhhhhhhhhhfgfhhgfhhhhgga^^ … … …
 Terminal Unix. Naviguer dans le dossier qui contient le fichier. Taper la commande more <nom_du_fichier> lbcd-05:GKG13demo deepseq$ more TNGGAACTTCATACCGTGCTCTCTGTAGGCACCATCAA. +HWIEAS210R_0028:2:1:3019:1114#AGAAGA/1. TNCTTGGACTACATATGGTTGAGGGTTGTACTGTAGGC. +HWIEAS210R_0028:2:1:3925:1114#AGAAGA/1. +HWIEAS210R_0028:2:1:6220:1114#AGAAGA/1. +HWIEAS210R_0028:2:1:6252:1115#AGAAGA/1. TNAATGCACTATCTGGTACGACTGTAGGCACCATCAAT. +HWIEAS210R_0028:2:1:6534:1114#AGAAGA/1. GNGGACTGAAGTGGAGCTGTAGGCACCATCAATAGATC. +HWIEAS210R_0028:2:1:8869:1114#AGAAGA/1. aBaaaeeeeehhhhhhhhhhhhfgfhhgfhhhhgga^^ … … …")
7
Combien de séquences dans mon fichier ?
Terminal Unix. Naviguer dans le dossier qui contient le fichier Taper la commande wc - l <nom_du_fichier> lbcd-05:GKG13demo deepseq$ wc -l GKG-13.fastq GKG-13.fastq >>> / 4 séquences

8
Mes séquences contiennent-elles le bon adaptateur ?
Séquence de mon adaptateur: CTGTAGGCACCATCAAT Taper la commande cat <nom_du_fichier> | grep CTGTAGG | wc -l lbcd-05:GKG13demo deepseq$ wc -l GKG-13.fastq | grep CTGTAGG | wc -l sur séquences Pas mal A contrario lbcd-05:GKG13demo deepseq$ wc -l GKG-13.fastq | grep ATCTCGT| wc -l 308

9
Quelle est la qualité de mes séquences ?
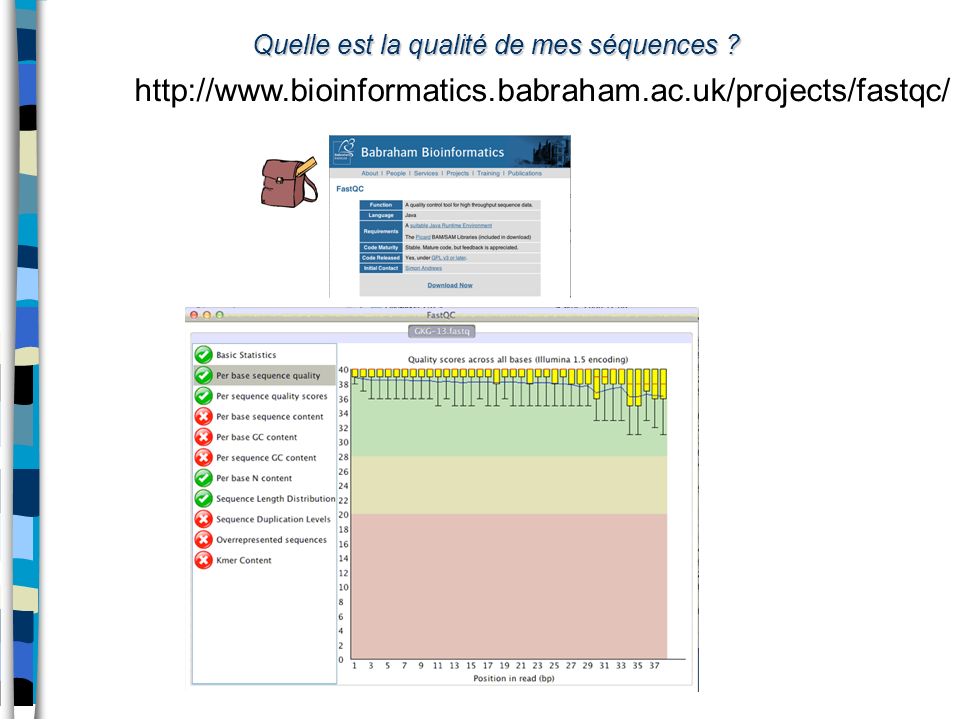
10
Comment retirer l’adaptateur ?
Séquence de mon adaptateur: CTGTAGGCACCATCAAT deepseq$ fastq_to_fasta -r –n -i GKG-13.fastq -o GKG-13.fasta deepseq$ more GKG-13.fasta >1 AATGGCACTGGAAGAATTCACCTGTAGGCACCATCAAT >2 TCTCGGTAGAACCTCCACTGTAGGCACCATCAATAGAT >3 TTTGTGACCGACACTAACGGGTACTGTAGGCACCATCA >4 TGGAATGTAAAGAAGTATGGAGCTGTAGGCACCATCAA >5 GTCAGCAACTTGATTCCAGCAATCTGTAGGCACCATCA >6 AATGGCACTGGAAGAATTCACGGGCTGTAGGCACCATC >7 TGGAAGACTAGTGATTTTGTTCTGTAGGCACCATCAAT >8 TGAACACAGCTGGTGGTATCCCTGTAGGCACCATCAAT deepseq$ fastx_clipper -a CTGTAGGCACCATCAAT -l 18 -i GKG-13.fasta -o GKG-13_clipped.fasta >18 AATGGCACTGGAAGAATTCAC >20 TTTGTGACCGACACTAACGGGTA >21 TGGAATGTAAAGAAGTATGGAG >22 GTCAGCAACTTGATTCCAGCAAT >23 AATGGCACTGGAAGAATTCACGGG >24 TGGAAGACTAGTGATTTTGTT >25 TGAACACAGCTGGTGGTATCC >26 TAAGTACTAGTGCCGCAGGA >27 TGAACACAGCTGGTGGTATC >28 TAGGAACTTCATACCGTGCTCT deepseq$ more GKG-13_clipped.fasta deepseq$ fastq_to_fasta -r -n -i GKG-13.fastq | fastx_clipper -a CTGTAGGCACCATCAAT -l 18 -o GKG-13_clip-pipe.fasta

11
J’utilise fastx_clipper et fastQC pour visualiser la distribution de taille de mes séquences
deepseq$ fastx_clipper -a CTGTAGGCACCATCAAT -l 0 -i GKG-13.fastq -o GKG-13_clipped.fastq deepseq$ more GKG-13_clipped.fastq @HWIEAS210R_0028:2:1:1313:1120#AGAAGA/1 AATGGCACTGGAAGAATTCAC +HWIEAS210R_0028:2:1:1313:1120#AGAAGA/1 fe\gggd\fgeeeggdaggag @HWIEAS210R_0028:2:1:1387:1119#AGAAGA/1 TCTCGGTAGAACCTCCA +HWIEAS210R_0028:2:1:1387:1119#AGAAGA/1 gggggeggfffgggfff @HWIEAS210R_0028:2:1:1849:1120#AGAAGA/1 TTTGTGACCGACACTAACGGGTA +HWIEAS210R_0028:2:1:1849:1120#AGAAGA/1 hhhhhhhhhfhgfhhhhgehhha

12
Je télécharge Bowtie, je l’installe, et je lis le manuel
Bowtie aligne des reads sur un génome de référence préalablement préparé Je télécharge Bowtie, je l’installe, et je lis le manuel Je télécharge mon génome au format FASTA Je prépare mon « index » Bowtie deepseq$ bowtie-build fasta_libraries/dmel-all-chromosome-r5.37.fasta dmel-r5.37 ~5 min deepseq$ ls –laht -rw-r--r deepseq staff M Mar 24 17:24 dmel-r5.37.rev.1.ebwt -rw-r--r deepseq staff M Mar 24 17:24 dmel-r5.37.rev.2.ebwt -rw-r--r deepseq staff M Mar 24 17:20 dmel-r ebwt -rw-r--r deepseq staff M Mar 24 17:20 dmel-r ebwt -rw-r--r deepseq staff K Mar 24 17:16 dmel-r ebwt -rw-r--r deepseq staff M Mar 24 17:16 dmel-r ebwt

13
J’aligne mes reads avec bowtie
deepseq$ bowtie ~/bin/bowtie/indexes/5.43_Dmel/5.43_Dmel -f GKG-13_clip-pipe.fasta -v 1 -k 1 -p 12 --al droso_matched_GKG-13.fa --un unmatched_GKG13.fa > GKG13_bowtie_output.tabulated ~/bin/bowtie/indexes/5.43_Dmel/5.43_Dmel -f GKG-13_clip-pipe.fasta -v 1 -k 1 -p 12 --al droso_matched_GKG-13.fa --un unmatched_GKG13.fa > GKG13_bowtie_output.tabulated # reads processed: # reads with at least one reported alignment: (84.12%) # reads that failed to align: (15.88%) Reported alignments to 1 output stream(s)
 # reads that failed to align: (15.88%) Reported alignments to 1 output stream(s)")
14
… et je récupère Un fichier d’alignement
deepseq$ ls -laht -rw-r--r deepseq staff 351M Mar 24 17:46 GKG13_bowtie_output.tabulated -rw-r--r deepseq staff 156M Mar 24 17:46 droso_matched_GKG-13.fa -rw-r--r deepseq staff 28M Mar 24 17:46 unmatched_GKG13.fa Un fichier d’alignement deepseq$ more GKG13_bowtie_output.tabulated L TGGAATGTAAAGAAGTATGGAG L GTGAATTCTCCCAGTGCCAAG R TGAACACAGCTGGTGGTATCC L CCCGTGAATTCTTCCAGTGCCATT R TGAACACAGCTGGTGGTATC R TCCTGCGGCACTAGTACTTA L GTGAATTCTTCCAGTGCCATT R ATTGCTGGAATCAAGTTGCTGAC L TTTGTGACCGACACTAACGGGTA R TGGAAGACTAGTGATTTTGTT L TAGGAACTTCATACCGTGCTCT X CTTGTGCGTGTGACAGCGGCT RHet TGGCGACCGTGACAGGACCCG R TGAACACAGCTGGTGGTATCC Un fichier des séquences alignées Un fichier des séquences non alignées deepseq$ more droso_matched_GKG-13.fa >21 TGGAATGTAAAGAAGTATGGAG >26 TAAGTACTAGTGCCGCAGGA >24 TGGAAGACTAGTGATTTTGTT >23 AATGGCACTGGAAGAATTCACGGG >27 TGAACACAGCTGGTGGTATC deepseq$ more unmatched_GKG13.fa >29 AGGGGGCTATTTCACTACTGGA >33 CGATGATGACGGTACCCGTAGA >37 GCTAGTCGGTACTTGAAAC >59 TGGTTGCAATAGCTTCTGGCGGA >61 GATGAGTGCTAGATGTAGGGA

15
Un pipeline d’annotations « génomiques »
Sequence reads (fasta format) Bowtie Pre-miRNAs (miRBase) Matched reads (fasta) Read Count Unmatched reads Bowtie Non coding RNAs Matched reads (fasta) Read Count Unmatched reads hierarchical annotation of sequence datasets Bowtie Transposons Matched reads (fasta) Read Count Unmatched reads Bowtie Genes Matched reads (fasta) Read Count Unmatched reads Bowtie Intergenic regions Matched reads (fasta) Read Count Unmatched reads Bowtie Viruses, transgenes, etc… Matched reads (fasta) Read Count Remaining unmatched sequences
 Bowtie. Pre-miRNAs (miRBase) Matched reads. (fasta) Read Count. Unmatched reads. Bowtie. Non coding RNAs. Matched reads. (fasta) Read Count. Unmatched reads. hierarchical. annotation. of. sequence. datasets. Bowtie. Transposons. Matched reads. (fasta) Read Count. Unmatched reads. Bowtie. Genes. Matched reads. (fasta) Read Count. Unmatched reads. Bowtie. Intergenic regions. Matched reads. (fasta) Read Count. Unmatched reads. Bowtie. Viruses, transgenes, etc… Matched reads. (fasta) Read Count. Remaining unmatched sequences.")
16
Je veux visualiser mes reads dans un « Genome Browser »
Un pipeline sommaire pour préparer un fichier de visualisation deepseq$ bowtie -v 1 -M 1 --best /Users/deepseq/bin/bowtie/indexes/5.37_Dmel -p 12 -f GKG-13_clip-pipe.fasta -S | samtools view -bS -o GKG-13_clip-pipe.fasta.bam - ; samtools sort GKG-13_clip-pipe.fasta.bam GKG-13_clip-pipe.fasta.bam.sorted ; samtools index GKG-13_clip-pipe.fasta.bam.sorted.bam 306K GKG-13_clip-pipe.fasta.bam.sorted.bam.bai 42M GKG-13_clip-pipe.fasta.bam.sorted.bam 80M GKG-13_clip-pipe.fasta.bam

17
Je veux visualiser mes reads dans un « Genome Browser » (2)
J’upload mes fichiers bam et bai sur un serveur accessible J’indique l’URL du fichier bam à Ensembl (Gbrowse, Modencode, etc..)
")
18
Je veux visualiser mes reads dans un « Genome Browser » (3)
Je navigue dans les régions d’intérêt, après avoir indiqué au Browser d’inclure mon « track »

19
Je veux visualiser mes reads dans un « Genome Browser » (4)
Encore un…

20
Un “profiler” maison pour les micros ARNs
deepseq$ miRNA_bowtie_profiler.py GKG-13_clip-pipe.fasta ~/bin/bowtie/indexes/dme_miR_r17.1.ebwt Sequence reads (fasta format) Bowtie Pre-miRNAs (miRBase) Indéxé pour Bowtie Bowtie Output Analyse “textuelle” Cartes des reads par miRNA Liste de comptage par miR_5p et miR_3p # bowtie -v 1 -M 1 --best --strata -p 12 --norc --suppress 2,6,7,8 /Users/deepseq/bin/bowtie/indexes/dme_miR_r17 -f GKG-13_clip-pipe.fasta # reads processed: # reads with at least one reported alignment: (64.81%) # reads that failed to align: (34.36%) # reads with alignments sampled due to -M: (0.84%) Reported alignments to 1 output stream(s) # Parsing completed in 1 minutes and 36.7 seconds
 Bowtie. Pre-miRNAs (miRBase) Indéxé pour Bowtie. Bowtie Output. Analyse textuelle Cartes des reads par miRNA. Liste de comptage par miR_5p et miR_3p. # bowtie -v 1 -M 1 --best --strata -p 12 --norc --suppress 2,6,7,8 /Users/deepseq/bin/bowtie/indexes/dme_miR_r17 -f GKG-13_clip-pipe.fasta. # reads processed: # reads with at least one reported alignment: (64.81%) # reads that failed to align: (34.36%) # reads with alignments sampled due to -M: (0.84%) Reported alignments to 1 output stream(s) # Parsing completed in 1 minutes and 36.7 seconds.")
21
miRNA_bowtie_profiler.py : Cartes des reads, par miR
offsets counts sizes

22
miRNA_bowtie_profiler.py : Attribution des reads “5p” et “3p”
miRs « 5p » miRs « 3p » + 987 reads 16003 reads = 16990, ~ reads *

23
miRNA_bowtie_profiler.py : Liste de comptage des miRs

24
Analyse d’expression différentielle
deepseq$ miRNA_bowtie_profiler.py GKG-13_clip-pipe.fasta ~/bin/bowtie/indexes/dme_miR_r17.1.ebwt Sequence reads (fasta format) Pre-miRNAs (miRBase) Indéxé pour Bowtie Bowtie Bowtie Output Analyse “textuelle” Cartes des reads par miRNA Liste de comptage par miR_5p et miR_3p DESeq Heatplus edgeR (Bioconductor)
 Pre-miRNAs (miRBase) Indéxé pour Bowtie. Bowtie. Bowtie Output. Analyse textuelle Cartes des reads par miRNA. Liste de comptage par miR_5p et miR_3p. DESeq. Heatplus. edgeR. (Bioconductor)")
25
Profiling des miRNAs durant la métamorphose de la drosophile
days L3 PF PF+12h Ecdysone titer Read count table

26
Clustering of miRNA read counts after normalization
days L3 PF PF+12h Clustering of miRNA read counts after normalization Ecdysone titer DESeq Heatplus

27
Analyse d’expression différentielle
PF PF+12h Larva « Differential calling » avec le jeu complet de données Metamorphosis Up-regulated 27 Down_regulated « Differential calling » sans replicats Metamorphosis Up-regulated Down_regulated Message: Le Deep Seq n’échappe pas au tests statistiques Les réplicats sont nécessaires pour estimer le bruit biologique

28
Naive and primed murine pluripotent stem cells have distinct miRNA signatures
ESC1 ESC2 EpiSC2 EpiSC1 EpiSC3 miR miR-302/367 miR17-92 M. Cohen-Tannoudji (Institut Pasteur) Jouneau (INRA Jouy en Josas) E. Heard (Institut Curie) C. Antoniewski (Institut Pasteur) 28/40
 Jouneau (INRA Jouy en Josas) E. Heard (Institut Curie) C. Antoniewski (Institut Pasteur) 28/40.")
29
Normalized miR read count profiles
29/40

30
A lattice of miR read profiles for rapid, visual annotation
30/40

31
31/40

32
“Stereo” lattice reveals changes in miR biogenesis between ES and EpiSCs
% length ESC EpiSC 32/40

33
Small RNA signatures AUGCUUUCAUGGCAUCCUUAC UUUACGAAAGUACCGUAGGAA -100
+100 |||||||||||||||||||||

34
Signature piRNA P-element Cartographie des ARN de 24-26nt d’ovaires de drosophile UUGCUUUCAUGGCAUCCUUACCGAUC AGCUUCUUUACGAAACGAAAGUACCG -100 ||||||||||||||||||||| +100

35
Signature piRNA P-element Cartographie des ARN de 24-26nt d’ovaires de drosophile UUGCUUUCAUGGCAUCCUUACCGAUC AGCUUCUUUACGAAACGAAAGUACCG -100 ||||||||||||||||||||| +100

Présentations similaires





![[number 1-100].](/1/172887/big_thumb.jpg "[number 1-100].>")











 et dénombrer (Entoure dans la bande numérique.>")

