Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
BioInformatique des micropuces
Abdoulaye Baniré Diallo 25 mars 2008

2
plan Introduction Types de puces Déroulement d’une expérience
Mise au point d’une puce Choix des gènes et des sondes Analyse des données d’expression Analyse des images et segmentation Normalisation

3
Plan(2) Analyse des données d’expression Types d’analyse Clustering
Comparaison de profils Différentes types de distances classificateur

4
Applications Identifier des gènes différentiellement exprimés (sur ou sous exprimés) dans des conditions déterminées: Maladie ou traitement Réponse à un stress ou à un signal Déterminer un profil d’expression lié à un état particulier de la cellule: Classification des sous types de cancer Déterminer tout ou une partie du réseau de régulation: Gènes d’expression similaires (recherche de promoteurs communs) Réseau de régulation
 Réseau de régulation.")
5
Format d’une micropuce
Spot: ensemble de Sondes spéciques à une cible (un gène par exemple) Sonde: une séquence de nucléotides
 Sonde: une séquence de nucléotides.")
6
Hybridation

7
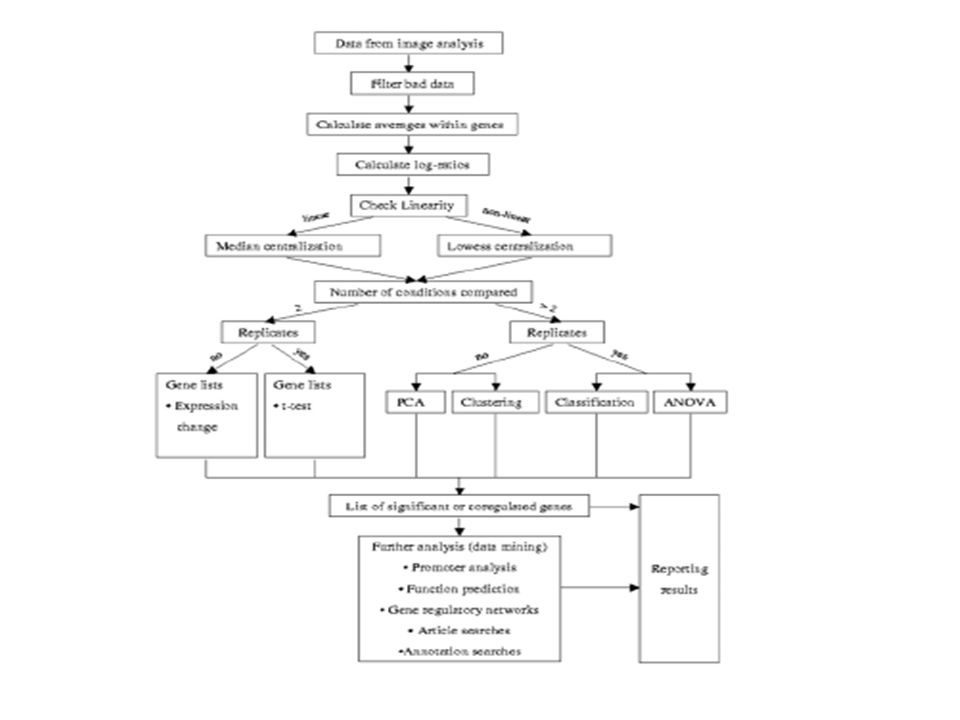
Réalisation d’une analyse
Une image vaut 1000 mots et un ensemble d’images animées?

8
Résumé

9
Étape de réalisation d’une analyse

10
cDNA microArray

11
Oligonucléotides genechips (Affymetrix)
")
12
Oligonucléotides genechips (Affymetrix)
")
13
Oligonucléotides genechips (Affymetrix)
Perfect Match: G1,…,Gk: k spots, 2 à 2 différents, spécifiques au gène G MisMatch: sondes avec une modification au milieu de la séquence. Capture les hybridations non spécifiques

14
complément Principale différence: Genechip
Micropuce = expression différentielle Genechip = expression pour un seul échantillon Genechip 2 types de redondance Spots multiples et différents pour un seul gène PM vs MM

15
Déroulement d’une expérience
Mise au point de la puce Choix du type de puce (oligos, cDNA,…) Sélection des gènes à mésurer Sélection des sondes Manufacture ou commande de la puce Utilisation Hybridation - lavage Analyse Mesure de l’expression de chaque gène (image- matrice numérique) Analyse statistique (normalisation, validation) Interprétation des résultats (clustering, data mining…) Gestion des données
 Sélection des gènes à mésurer. Sélection des sondes. Manufacture ou commande de la puce. Utilisation. Hybridation - lavage. Analyse. Mesure de l’expression de chaque gène (image- matrice numérique) Analyse statistique (normalisation, validation) Interprétation des résultats (clustering, data mining…) Gestion des données.")
17
Mise au point d’une puce
Choix des gènes à mesurer Choix des sondes Acheter tout fait Récupérer sur le web Geo (NCBI), NWG, Affymetrix,… La mettre au point soi-même Où récupérer les gènes? Comment choisir les sondes?
, NWG, Affymetrix,… La mettre au point soi-même. Où récupérer les gènes Comment choisir les sondes")
18
Gènes Informations à conserver Bases de données UniGene (NCBI) TIGR
Regrouperment (clusters) d’ARNm et EST de Genbank (1 gène par groupe) TIGR Même principe qu’UniGene REFSEQ (NCBI) Banque de séquences de qualité grande Ucsc genome browser Banque de données de diverses informations génomiques de l’humain
 d’ARNm et EST de Genbank (1 gène par groupe) TIGR. Même principe qu’UniGene. REFSEQ (NCBI) Banque de séquences de qualité grande. Ucsc genome browser. Banque de données de diverses informations génomiques de l’humain.")
19
Banques de données Unigène – décembre 2007 TIGR – février 2008
66488 groupes (>> 30000)!!, mRNA séquences au total Contient de nombreuses informations (tissus, NCBI, lignée,…) Manque: séquences consensus, épissage, stabilité TIGR – février 2008 Plus de cluster et plus de 5 millions d’EST Séquence consensus, ontologie du gène, épissage alternatif, réseau métabolique RefSeq – Donne des références stables sur l’identification, la caractérisation, les analyses de mutations, les études d’expression … Utilise des numéros d’accession et peu de séquences (environ ) Uscs Genome browser Répertorie les analyses au niveau génomique effectuées Prédiction de gènes, expression, régulation Utile dans le cadre des tiling arrays
!!, mRNA séquences au total. Contient de nombreuses informations (tissus, NCBI, lignée,…) Manque: séquences consensus, épissage, stabilité. TIGR – février Plus de cluster et plus de 5 millions d’EST. Séquence consensus, ontologie du gène, épissage alternatif, réseau métabolique. RefSeq – Donne des références stables sur l’identification, la caractérisation, les analyses de mutations, les études d’expression … Utilise des numéros d’accession et peu de séquences (environ ) Uscs Genome browser. Répertorie les analyses au niveau génomique effectuées. Prédiction de gènes, expression, régulation. Utile dans le cadre des tiling arrays.")
20
Choix des sondes ou primers
3 conditions Sensibilité Une bonne sonde "hybride" hybride bien avec sa cible et produit un signal représentant son niveau d’expression Spécificité Une bonne sonde n’hybride pas avec une d’autres cibles (cross hybridization) Comportement isothermal Chaque sonde "hybride" avec sa cible à une température optimale L’intervalle des températures de toutes n’est pas trop large Comment combiner ces conditions? Autant de stratégies que de logiciels
 Comportement isothermal. Chaque sonde hybride avec sa cible à une température optimale. L’intervalle des températures de toutes n’est pas trop large. Comment combiner ces conditions Autant de stratégies que de logiciels.")
21
Choix des sondes Sensibilité: éviter les repliements en structure secondaire stable Calcul du repliement optimal: MFOLD, Rnafold Spécificité: éviter qu’une partie de la sonde soit similaire à plusieurs gènes: Blast Position dans le transcrit: plus on est proche du début de la reverse transcriptase (fin 3’) mieux c’est Température: Différentes techniques de calcul de la température de fusion (hybridation)
 mieux c’est. Température: Différentes techniques de calcul de la température de fusion (hybridation)")
22
Spécificité d’une sonde
Nettoyage ou filtrage Nous voulons éviter les séquences Ambigues (mauvais séquençage) De faible complexité Longues séquences de nucléotides identiques: TTTTT…TT Répétitions: TATATA…TA Séquences communes à plusieurs gènes Contaminant, vecteurs !!!!!! Outils RepeatMasker (RepBase) MaskerAid (WuBlast) OligoArray Dust (Blast) Important: séquences non nettoyées créent plusieurs problèmes à l’expériences
 De faible complexité. Longues séquences de nucléotides identiques: TTTTT…TT. Répétitions: TATATA…TA. Séquences communes à plusieurs gènes. Contaminant, vecteurs !!!!!! Outils. RepeatMasker (RepBase) MaskerAid (WuBlast) OligoArray. Dust (Blast) Important: séquences non nettoyées créent plusieurs problèmes à l’expériences.")
23
Calcul de la température de fusion
Température de fusion = température à laquelle 50% d’une sonde s’hybride avec son brin complémentaire Paramètres importants Nucléotides de la sonde Concentration C du brin (inconnu en général) Concentration en sodium (Na+) de la solution contenant les cibles Une équation simple pour des oligos assez long (> 50 nt) utilisé par Qiagen pour des oligos de 70mer
 Concentration en sodium (Na+) de la solution contenant les cibles. Une équation simple pour des oligos assez long (> 50 nt) utilisé par Qiagen pour des oligos de 70mer.")
24
Calcul de la température : modèle NN
Formule de base Tm = H /(S – R ln (C/4)) H = enthalpie (chaleur absorbée par la création du lien G-C) S = entropie ("perte de dynamisme") Modèle NN (nearest-neighbor) 5’ TAACCACGAT | | | fermeture ATTGGTGCTA
) H = enthalpie (chaleur absorbée par la création du lien G-C) S = entropie ( perte de dynamisme ) Modèle NN (nearest-neighbor) 5’ TAACCACGAT. | | | fermeture. ATTGGTGCTA.")
25
SantaLucia et al. 1998

26
Spécificité: hybridation croisée
Le problème n séquences S1…Sn l = longueur de sonde Trouver n sous séquences P1…Pn tel que: Pour tout i /= j Pi n’est pas "similaire" à une sous séquence de Sj Similarité Blast

27
Choix des sondes et règles de sélection
Une fois un gène est choisie dans une analyse, une sonde est choisie pour lui avec un ensemble optimal de paramètres (un grand nombre de 70mer candidats) Tous les oligos sont entre 78°C± 5°C en utilsant la formule Où [Na+] = 0.1M et length = #A + #C + #G + #T Chaque oligo est autour de 1000 bases de 3’ end de la séquence disponible Un oligo ne peut avoir une répétition contigue d’un seul nucléotide (ou poly(N)) de plus de 7 bases
 Tous les oligos sont entre 78°C± 5°C en utilsant la formule. Où [Na+] = 0.1M et length = #A + #C + #G + #T. Chaque oligo est autour de 1000 bases de 3’ end de la séquence disponible. Un oligo ne peut avoir une répétition contigue d’un seul nucléotide (ou poly(N)) de plus de 7 bases.")
28
Choix des sondes et règles de sélection
Une oligo ne peut avoir une potentielle hairpin avec une tige de plus de 9 bases Un score normalisé est assigné à chaque oligo basé sur le nombre de répétitions Les oligos avec plus de répétitions ayant un score plus grand que le seuil sont filtrés

29
Choix des sondes et règles de sélection
Chaque Oligo a un score <= 70% d’identité avec tous les autres gènes Utilisez Blast sur les séquences de l’humain Chaque Oligo de n’importe quelle taille ne peut avoir plus de 20 bases communes contigues avec n’importe quelle autre gène Au final: Une fois que les candidats ont été choisies, les oligos sont choisis avec un score minimal de blast (cross-hybridization)
")
30
Oligo Array 1.0 SCANNER PAGE

31
Analyse de données d’expression
Données d’expression n gènes et m échantillons (puces) Expressions normalisées sur chaque puce et entre les puces Gènes: certaines valeurs d’expression peuvent manquer Samples/puces: Patients (sains/malades) Expérience temporelle (ei,1; …;ei,m) = profil d’expression du gène i (e1,j; …;en,j) = profil d’expression d’un échantillon
 Expressions normalisées sur chaque puce et entre les puces. Gènes: certaines valeurs d’expression peuvent manquer. Samples/puces: Patients (sains/malades) Expérience temporelle. (ei,1; …;ei,m) = profil d’expression du gène i. (e1,j; …;en,j) = profil d’expression d’un échantillon.")
32
Micropuces et analyse d’images
Table des intensités Gene 1: rouge 100 vert 125 …. 2 images (intensité rouge et intensité vert) (format Tiff) Combinaison d’analyses 3 problèmes Associer les pixels correspondant à un gène Calculer l’intensité Évaluer la qualité de la mesure
 (format Tiff) Combinaison d’analyses. 3 problèmes. Associer les pixels correspondant à un gène. Calculer l’intensité. Évaluer la qualité de la mesure.")
33
Association gène-pixels
Localisation des spots Structure micropuce n * m grilles 1 grille = k*l spots Problème Irrégularité du placement des spots Grilles non alignées Grilles courbées (verre) Espace entre grilles inconstant Spots inconstants dans une grille Doit être vérifié avant l’analyse
![]()
34
Association gène-pixels
Segmentation: différencier, dans la zone associée à un gène, les pixels présentant un signal dû à l’hybridation (foreground) du fond (background) Problème difficile de traitement d’image 4 méthodes (entre autre) Cercle fixe Cercle adaptatif Forme adaptative histogramme
![]()
35
Méthodes de segmentation
Cercle fixe (ScanAnalyze) Pas d’intervention utilisateur Méthode sommaire et limitée Cercle adaptatif Diamètre spécifique à chaque spot ScanAlyze: ajustable à la main Forme adaptative Non circulaire Algorithme de Watershed Étendre la zone « foreground" à partir d’un pixel de départ (seed) histogramme
 Pas d’intervention utilisateur. Méthode sommaire et limitée. Cercle adaptatif. Diamètre spécifique à chaque spot. ScanAlyze: ajustable à la main. Forme adaptative. Non circulaire. Algorithme de Watershed. Étendre la zone « foreground à partir d’un pixel de départ (seed) histogramme.")
36
Détermination du fond Nous avons tous les éléments pour transformer notre image en données numériques avant d’évaluer la qualité

37
Informations calculées
Foreground (signal) : ration Rouge/vert Moyenne et médian des pixels du signal La médiane est moins sensible aux pixels extrêmes Background idem Intensité (intégral, moyenne, mediane) et forme du signal Étiquette sur les pixels douteux (contrôle de la qualité) Signal moins fort que le fond Déviation standard élevée Signal trop bruité,…
 : ration Rouge/vert. Moyenne et médian des pixels du signal. La médiane est moins sensible aux pixels extrêmes. Background. idem. Intensité (intégral, moyenne, mediane) et forme du signal. Étiquette sur les pixels douteux (contrôle de la qualité) Signal moins fort que le fond. Déviation standard élevée. Signal trop bruité,…")
38
Exemple ImaGene

39
Résultat d’analyse d’expression

41
Saturation

42
Saturation Les spots partiellement saturés peuvent être traités en supprimant seulement les pixels aux alentours du spot Peut être réalisé par traitement d’image, un facteur de saturation de mois que 1 sera considéré Facteur de saturation = fraction des bons pixels non saturés Les spots complètements saturés ne peuvent être utilisés pour une analyse quantitative
![]()
43
Contrôle de la qualité Le contrôle de qualité d’un spot peut se faire par traitement d’image Score de QC = Aire /Perimètre Cercle idéal = R/2 Si score < cercle idéal, mauvaise forme Spot pixel > 2*median(bkg) est pris comme estimé du ratio signal/bruit > 50%
 est pris comme estimé du ratio signal/bruit > 50%")
44
Normalisation But: comparer les expressions de chaque spot pour déterminer les gènes sur ou sous exprimés Il faut que les mésures soient comparables Problèmes: Les expériences de micropuces sont soumises à de multiples biais aléatoires ou systématiques Données bruitées Résultats bruts non comparables souhait: éliminer les variations non biologiques pour qu’un gène qui est reconnu comme exprimé différentiellement le soit pour les raisons biologiques étudiées

45
Sources d’erreurs Aléatoires Systématiques Problème de correction
ARN hybridé: quantité ou préparation Conditions expérimentales ou qualité de la puce Puces multiples Biais spatiaux ou biais de couleurs Problème de correction Variation locale, intensité, non linéaire Pour diminuer les variations non corrigées: Réplicats biologiques: plusieurs puces (coût), samples pooling Réplicats techniques: sur une même puce ou différentes puces
, samples pooling. Réplicats techniques: sur une même puce ou différentes puces.")
46
Techniques de normalisation: survol
Interne à une puce (Rouge vs vert) MA-plot: M = log(R/V) A = log (sqrt(RV)) y- M: log ratio x-A: average log-intensity Si il n’y a pas de biais, on a en gros des données distribuées en nuage autour de y = 0 Principe de normalisation M = M-quelque chose Calculée sur un ensemble S de spots
 MA-plot: M = log(R/V) 1 A = log (sqrt(RV)) y- M: log ratio. x-A: average log-intensity. Si il n’y a pas de biais, on a en gros des données distribuées en nuage autour de y = 0. Principe de normalisation. M = M-quelque chose. Calculée sur un ensemble S de spots.")
47
Normalisation dépendant de l’intensité
M = M – Cs(A) Cs(A) = (h*A + c) h = pente c = décalage (h*A + c) = régression linéaire D’autres formes: non linéaire (Loess)
 Cs(A) = (h*A + c) h = pente. c = décalage. (h*A + c) = régression linéaire. D’autres formes: non linéaire (Loess)")
48
Régression linéaire

49
Régression non linéaire (Loess)
")
50
Normalisation entre plusieurs puces
Exemple: nous disposons d’une puce par patient et nous voulons comparer toutes les puces Technique Analyse de box plot Modification par Rééchelonnage Recentrage Normalisation de la distribution Hypothèse: Les variations proviennent du processus expérimental et non pas de valeur biologique

51
Normalisation entre plusieurs puces

52
Types d’analyse Recherche de groupes de gènes ayant des profils d’expression similaires Gènes réagissant de la même façon à un stimulus (froid, maladie, …) Recherche d’échantillons au profil similaire Classification des sous-types d’une maladie Solution: clustering Construction d’un classifieur ou prédicteur Diagnostic à partir du profil Inférence de réseaux de régulation
 Recherche d’échantillons au profil similaire. Classification des sous-types d’une maladie. Solution: clustering. Construction d’un classifieur ou prédicteur. Diagnostic à partir du profil. Inférence de réseaux de régulation.")
53
Comparaison entre 2 profils
Problème 2 vecteurs V1 et V2: profils V1 = (x1,…,xk) V2 = (y1,…,yk) (k = n ou m) Ces deux profils sont –ils similaires? Similarité ? Correlation distance
 V2 = (y1,…,yk) (k = n ou m) Ces deux profils sont –ils similaires Similarité Correlation. distance.")
54
Correlation R(V1,V2) = coefficient de corrélation entre les profils V1et V2 Représente le niveau de relation entre ces 2 profils -1 ≤ r ≤ 1 1: correlation positive -1: correlation négative 0: pas de correlation Mésure de colinéarité

55
Correlation standard (Pearson)
V1 = (1, 2, 3, 4) et V2 = (1, 2, 3, 4) => r = 1 V1 = (1, 2, 3, 4) et V2 = (4, 3, 2, 1) => r = -1 V1 = (1, 1, 1, 1) et V2 = (1, 1, 1, 1) => r = 0 Remarque: si les données ont été centrées avec moyenne 0 et écart type 1 alors R = somme des xiyi
 et V2 = (1, 2, 3, 4) => r = 1. V1 = (1, 2, 3, 4) et V2 = (4, 3, 2, 1) => r = -1. V1 = (1, 1, 1, 1) et V2 = (1, 1, 1, 1) => r = 0. Remarque: si les données ont été centrées avec moyenne 0 et écart type 1 alors. R = somme des xiyi.")
56
Correlation de pearson
Fortement correlé (r = 0.97)
")
57
Correlation de pearson
Correlé négativement (r = -0.47)
")
58
Corrélation de Spearman
Principe: prendre en compte l’ordre des xi et yi plutôt que leurs valeurs Exemple: V1 = (-4, 1, -2, 1) et V2 = (-3, -2, 1, -1) V1 = (1, 3, 2, 3) et V2 = (1, 2, 4, 3) But: minimiser l’influence du bruit et des outliers Plus spécifique que Pearson Moins sensible que Pearson Problème: Perte de la direction de la régulation
 et V2 = (-3, -2, 1, -1) V1 = (1, 3, 2, 3) et V2 = (1, 2, 4, 3) But: minimiser l’influence du bruit et des outliers. Plus spécifique que Pearson. Moins sensible que Pearson. Problème: Perte de la direction de la régulation.")
59
Mauvaise correlation due à un outlier
Correlation de 0.63 à cause de l’outlier

60
Corrélation et Jacknife
Principe: Éviter d’être trop sensible à un ou des outliers l = entier fixé (petit) pour le nombre d’outliers à éliminer au maximum En prenant le min, on élimine les cas où un ou plusieurs points sont dominants
 pour le nombre d’outliers à éliminer au maximum. En prenant le min, on élimine les cas où un ou plusieurs points sont dominants.")
61
Les distances Distance euclidienne Distance de correlation
Distance de Manhattan Information mutuelle Et d’autres

62
Distance euclidienne La distance que nous avons tous appris au secondaire Distance entre les échantillons Peut être généralisé à N dimensions Chaque gène est une dimension. Donc pour n gènes, nous avons un espace à n-dimension

63
Distance euclidienne est sensible à l’échelle
Bien que les profils soient similaires, BUR6 est beaucoup plus régulé que IDH1 (a)Distance euclidienne = (b) les données ont été mise en échelle en divisant par l’écart type D = 0.88
Distance euclidienne = 5.8 (b) les données ont été mise en échelle en divisant par l’écart type D =")
64
Différences entre la distance euclidienne et la corrélation de Pearson

65
Distance de Manhattan Appelé également city block distance |∆x| + |∆y|

66
Information mutuelle Si nous connaissons quelques choses d’une variable aléatoire X, quelle information peut –elle nous donner pour la distribution de probabilité Y Basée sur l’entropie L’entropie se définie comme L’information mutuelle se définie comme Implanté dans le score TNOM (treshold number of misclassification) pour distinguer des gènes
 pour distinguer des gènes.")
67
Clustering hiérarchique
Principe similaire aux algorithmes de clustering vus en phylogénie comme NJ ou PGM. E D: matrice de distances clustering arbre

68
Clustering des données d’expression
Principe E: matrice n*m de données d’expression C1,…, Ck : k groupes de profils Dans un bon clustering 2 profils appartenant à la même classe sont similaires: homogénéité des clusters 2 profils de classes différentes ne sont pas similaires: Séparabilité des clusters

70
Clustering: aspects techniques
3 points Choix de la distance Choix des 2 clusters à regrouper En général les 2 clusters les plus proches (Eisen, 1998) Calcul de la distance du nouveau cluster aux autres clusters
 Calcul de la distance du nouveau cluster aux autres clusters.")
71
Chaînage (linkage) Single linkage
d(C1,C2) = distance entre leurs éléments les plus proches Average linkage d(C1,C2) = moyenne des distances entre les gènes de chaque cluster Complete linkage d(C1,C2) = distance entre leurs éléments les plus éloignés Notes 3, 4 et 5
 = distance entre leurs éléments les plus proches. Average linkage. d(C1,C2) = moyenne des distances entre les gènes de chaque cluster. Complete linkage. d(C1,C2) = distance entre leurs éléments les plus éloignés. Notes 3, 4 et 5.")
72
Analyse d’un clustering
Bootstrap Rééchantillonnage de colonnes, lignes ou cases Arbre consensus + scores de bootstrap Bootstrap paramétrique Intégrer les paramètres statistiques connus dans le rééchantillonnage (variabilité, distribution, …) Choix des clusters Se baser sur la longueur des branches Voir notes 1 et 2
 Choix des clusters. Se baser sur la longueur des branches. Voir notes 1 et 2.")
73
Clustering par partitionnement: k-mean
Principe Définir une notion de représentant pour un cluster: Moyenne (centroide) : (1,2,3) et (3,1,2) => (2,1.5,2.5) Fixer le nombre K de clusters voulus Algorithme Assigner aléatoirement chaque profil à un des k clusters Calculer le représentant de chaque cluster Pour chaque profil x: déplacer x dans le cluster dont le dont le représentant est le plus proche de x Si aucun profil n’a changé de cluster: arrêter Sinon retourner en 2.
 : (1,2,3) et (3,1,2) => (2,1.5,2.5) Fixer le nombre K de clusters voulus. Algorithme. Assigner aléatoirement chaque profil à un des k clusters. Calculer le représentant de chaque cluster. Pour chaque profil x: déplacer x dans le cluster dont le dont le représentant est le plus proche de x. Si aucun profil n’a changé de cluster: arrêter. Sinon retourner en 2.")
74
Clustering différent par rapport au nombre de cluster

75
Caractéristiques du k-mean
But: essayer de minimiser le critère suivant: Technique: Algorithme de machine learning (apprentissage HMM) Défauts: Sensibilité à la partition initiale Répéter plusieurs fois et faire un consensus Choix du paramètre k En essayer plusieurs Multidimensional scaling (MDS) Validation Homogénéité vs séparabilité bootstrap
 Défauts: Sensibilité à la partition initiale. Répéter plusieurs fois et faire un consensus. Choix du paramètre k. En essayer plusieurs. Multidimensional scaling (MDS) Validation. Homogénéité vs séparabilité. bootstrap.")
76
Données d’expression et classification Construction d’un classificateur

77
Principaux problèmes Choix des gènes pertinents vis-à-vis de la classification souhaitée Méthode d’apprentissage Apprentissage supervisé car on connaît la classe de chaque sample Méthode de classification Validation du prédicateur Notions importantes Séparabilité des données Linéaire/ non linéaire

78
Séparabilité; linéarité

79
Séparabilité; linéarité

80
Séparabilité; linéarité

81
K-nearest-neighbor Données: Algorithme m samples classifiés (A)
p gènes classificateurs s un nouveau sample: données d’expression pour les p gènes choisis Deux paramètres k et l Algorithme Examiner les k samples les plus proches de s Assigner à s la classe contenant le plus grand nombre de samples parmi ces k voisins, sauf si on en a moins de l auquel cas s est non classifié

82
K-nearest-neighbor Propriétés Remarque: Positives Négatives
Rapide Apprentissage trivial Non sensible à la linéarité Négatives Sensible aux données trompeuses Sensible à la distance choisie Sensible aux choix des p gènes Remarque: Souvent l = 0 alors on parle de N-N

83
Classification par centroïde
Données m samples classifiés en k classes p gènes classificateurs s nouveaux samples ( p gènes) Algorithme Calculer le centroïde de chacune des k classes Affecter s à la classe du plus proche centroïde Propriétés Rapide, sans apprentissage Sensible à la non linéarité des données
 Algorithme. Calculer le centroïde de chacune des k classes. Affecter s à la classe du plus proche centroïde. Propriétés. Rapide, sans apprentissage. Sensible à la non linéarité des données.")
84
Classification par centroïde

85
Autres approches Analyse par discriminant Linéaire (LDA)
S’applique à 2 samples seulement Séparation par un hyperplan (droite) minimisant la variance dans chaque classe et maximisant la variance entre classes Considère seulement les données linéairement séparables Réseau de neurones artificiels Support Vector Machines (version sophistiqué de LDA) Validation Similaire aux HMM Training set / test set Cross validation LOOCV (similaire à la validation croisée, mais un seul sample laissé)
 minimisant la variance dans chaque classe et maximisant la variance entre classes. Considère seulement les données linéairement séparables. Réseau de neurones artificiels. Support Vector Machines (version sophistiqué de LDA) Validation. Similaire aux HMM. Training set / test set. Cross validation. LOOCV (similaire à la validation croisée, mais un seul sample laissé)")
Présentations similaires


, A. Idbaih (1,2), A. Reyniès (3), S. Lair (2), J>")


>")















