Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Unité de Recherche en Biologie Moléculaire
Christophe Lambert Unité de Recherche en Biologie Moléculaire FUNDP Développement d’une méthode automatique fiable de modélisation de la structure tridimensionnelle des protéines par homologie et application au protéome de Brucella melitensis FUNDP, 26 septembre 2003, Namur

2
Plan Introduction Buts Développement de ESyPAliNN
Développement de ESyPred3D Base de données Brucella melitensis Conclusions / perspectives

3
Brucella sp. Génome de Brucella melitensis
Bactérie Gram négative (-proteo bactérie) Pathogène intracellulaire facultatif d’animaux (stérilité et avortement) et des humains (fièvre de Malte ou brucellose). Zoonose mondiale. 6(8?) espèces sont définies Les génomes de deux espèces sont séquencés (Brucella melitensis et Brucella suis) Génome de Brucella melitensis 2 chromosomes ( bp et bp) 3197 protéines déduites (fonction assignée par similarité: 2487)
 Pathogène intracellulaire facultatif d’animaux (stérilité et avortement) et des humains (fièvre de Malte ou brucellose). Zoonose mondiale. 6(8 ) espèces sont définies. Les génomes de deux espèces sont séquencés (Brucella melitensis et Brucella suis) Génome de Brucella melitensis. 2 chromosomes ( bp et bp) 3197 protéines déduites (fonction assignée par similarité: 2487)")
4
Protéines Grande partie du poids sec des êtres vivants
Hétéropolymères d’acides aminés valine (V) leucine (L) méthionine (M) phénylalanine (F) tyrosine (Y) isoleucine (I) tryptophane (W) glycine (G)
 leucine (L) méthionine (M) phénylalanine (F) tyrosine (Y) isoleucine (I) tryptophane (W) glycine (G)")
5
Introduction Structure 3D: information importante pour
mieux comprendre la fonction d’une protéine les interactions avec des ligands ou d’autres protéines planifier de la mutagenèse dirigée Nombre de structures connues (~15.000) est plus petit que le nombre de séquences connues (~ ) Techniques expérimentales: longues et coûteuses Alternative: modélisation
 est plus petit que le nombre de séquences connues (~ ) Techniques expérimentales: longues et coûteuses. Alternative: modélisation.")
6
Modélisation par homologie ou comparative modeling
Recherche en banque de données PDB template Etape critique Construction du modèle 3D Evaluation du modèle Alignement Cible-template

7
MAO B (template: 1f8r, LAAO)
2 MAO B (PDB ID: 1gos)
")
8
Modélisation par homologie
Recherche en banques de séquences de structures connues DIM1p (Saccharomyces cerevisiae) MGKAAKKKYSGATSSKQVSAEKHLSSVFKFNTDLGQHILKNPLVAQGIVDKAQIRPSDVVLEVGPGTGNL TVRILEQAKNVVAVEMDPRMAAELTKRVRGTPVEKKLEIMLGDFMKTELPYFDICISNTPYQISSPLVFK LINQPRPPRVSILMFQREFALRLLARPGDSLYCRLSANVQMWANVTHIMKVGKNNFRPPPQVESSVVRLE IKNPRPQVDYNEWDGLLRIVFVRKNRTISAGFKSTTVMDILEKNYKTFLAMNNEMVDDTKGSMHDVVKEK IDTVLKETDLGDKRAGKCDQNDFLRLLYAFHQVGIHF Score E Sequences producing significant alignments: (bits) Value pdb|1YUB Solution Structure Of An Rrna Methyltransferase e-09 pdb|1QAN Chain A, The Structure Of The Rrna Methyltransfe e-08 pdb|1G6Q Chain 1, Crystal Structure Of Yeast Arginine Met pdb|1EI1 Chain A, Dimerization Of E. Coli Dna Gyrase B Pr pdb|3HDH Chain A, Pig Heart Short Chain L-3-Hydroxyacyl C pdb|1PSZ Chain A, Pneumococcal Surface Antigen Psaa pdb|1VID Catechol O-Methyltransferase
 MGKAAKKKYSGATSSKQVSAEKHLSSVFKFNTDLGQHILKNPLVAQGIVDKAQIRPSDVVLEVGPGTGNL. TVRILEQAKNVVAVEMDPRMAAELTKRVRGTPVEKKLEIMLGDFMKTELPYFDICISNTPYQISSPLVFK. LINQPRPPRVSILMFQREFALRLLARPGDSLYCRLSANVQMWANVTHIMKVGKNNFRPPPQVESSVVRLE. IKNPRPQVDYNEWDGLLRIVFVRKNRTISAGFKSTTVMDILEKNYKTFLAMNNEMVDDTKGSMHDVVKEK. IDTVLKETDLGDKRAGKCDQNDFLRLLYAFHQVGIHF. Score E. Sequences producing significant alignments: (bits) Value. pdb|1YUB Solution Structure Of An Rrna Methyltransferase e-09. pdb|1QAN Chain A, The Structure Of The Rrna Methyltransfe e-08. pdb|1G6Q Chain 1, Crystal Structure Of Yeast Arginine Met pdb|1EI1 Chain A, Dimerization Of E. Coli Dna Gyrase B Pr pdb|3HDH Chain A, Pig Heart Short Chain L-3-Hydroxyacyl C pdb|1PSZ Chain A, Pneumococcal Surface Antigen Psaa pdb|1VID Catechol O-Methyltransferase")
9
Modélisation par homologie Alignement cible-template
DIM1p MGKAAKKKYSGATSSKQVSAEKHLSSVFKFNTDLGQHILKNPLVAQGIVDKAQIRPSDVV 1YUB MNKNIKYSQNFLTSEKVLNQIIKQLNLKETDTV DIM1p LEVGPGTGNLTVRILEQAKNVVAVEMDPRMAAELTKRVRGTPVEKKLEIMLGDFMKTELP 1YUB YEIGTGKGHLTTKLAKISKQVTSIELDSHLFNLSSEKLK---LNTRVTLIHQDILQFQFP ¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯¯ DIM1p YFD--ICISNTPYQISSPLVFKLINQPRPPRVSILMFQREFALRLLARPGDSLYCRLSAN 1YUB NKQRYKIVGNIPYHLSTQIIKKVVFESRASDI-YLIVEEGFYKRTLD-----IHRTLGLL DIM1p VQMWANVTHIMKVGKNNFRPPPQVESSVVRLEIKNPRPQVDYNEWDGLLRIVFVRKNRTI 1YUB LHTQVSIQQLLKLPAECFHPKPKVNSVLIKLTRHTTDVPDKY--WK--LYTYFVSK---- DIM1p SAGFKSTTVMDILEKNYKTFLAMNNEMVDDTKGSMHDVVKEKIDTVLKETDLGDKRAGKC 1YUB WVNREYRQLFTKN QFHQAMKHAKVNN--LSTI DIM1p DQNDFLRLLYAFHQVGIHF 1YUB TYEQVLSIFNSYLLFNGR- %id. = 18%

10
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... 1YUB Y E I G T G K G H L

11
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E I G T G K G H L

12
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E I G T G K G H L

13
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G T G K G H L

14
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G T G K G H L

15
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G P G K G H L

16
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G P G K G H L

17
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G P G T G H L

18
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G P G T G H L

19
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G P G T G N L

20
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... L E V G P G T G N L

21
Assignation des coordonnées
Modélisation par homologie Assignation des coordonnées DIM1p LEVGPGTGNLTVRILEQAKNV... 1YUB YEIGTGKGHLTTKLAKISKQV... Modèle de DIM1p L E V G P G T G N L

22
%id. 100 50 40 30 25 20 Modélisation par homologie (fiable)
Modélisation par homologie (fiable) Alignement pairé: la plupart des caractéristiques bien prédites Alignement multiple: beaucoup de caractéristiques bien prédites Twilight zone Combinaison d’alignements et données expérimentales quelques caractéristiques bien prédites Midnight zone reconnaissance de fold (pas très fiable) Protéines pas nécessairement homologues MAIS des protéines de séquences différentes peuvent adopter le même fold
 Alignement pairé: la plupart des caractéristiques bien prédites. Alignement multiple: beaucoup de caractéristiques bien prédites. Twilight zone. Combinaison d’alignements et données expérimentales. quelques caractéristiques bien prédites. Midnight zone. reconnaissance de fold (pas très fiable) Protéines pas nécessairement homologues MAIS des protéines de séquences différentes peuvent adopter le même fold.")
23
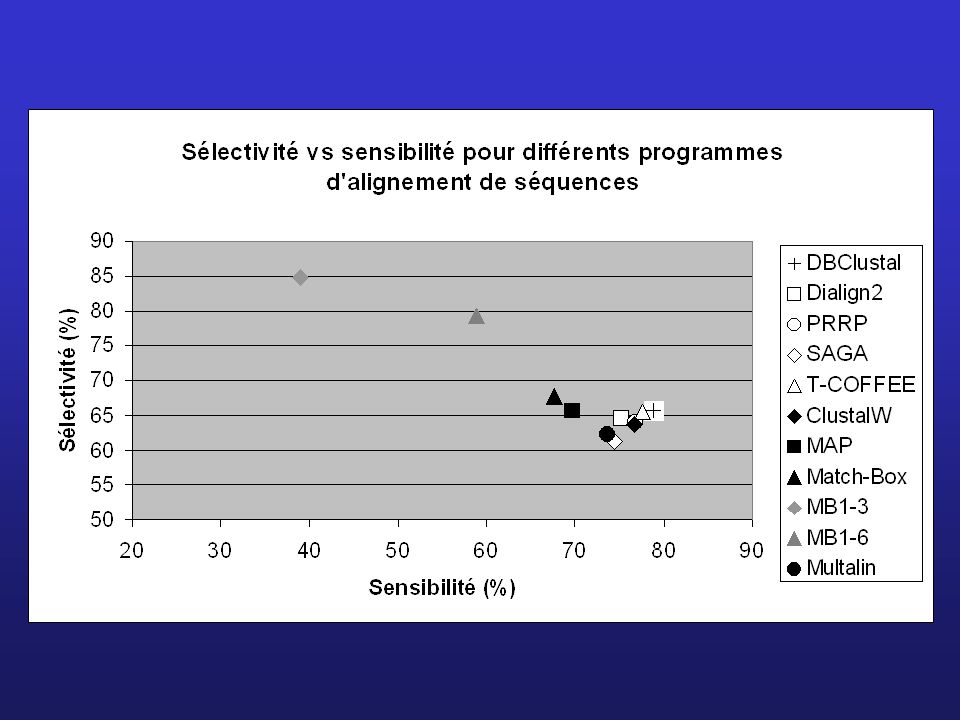
100 10/535 Sélectivité (%) 50 10 10 50 100 Sensibilité (%) 1/1 1/2
1/10 5/5 5/10 5/50 10/10 10/20 10/100 100 10/535 Sélectivité (%) 50 10 10 50 100 Sensibilité (%)
 Sensibilité (%)")
26
Plan Introduction Buts Développement de ESyPAliNN
Développement de ESyPred3D Base de données Brucella melitensis Conclusions / perspectives

27
Buts Développer une méthode fiable d’alignement pairé de séquences
Développer un programme de modélisation automatique par homologie Prédire la structure 3D des protéines déduites du génome de Brucella melitensis

28
Plan Introduction Buts Développement de ESyPAliNN
Développement de ESyPred3D Base de données Brucella melitensis Conclusions / perspectives

29
Limites de l’alignement multiple
Thompson J.D. et al. Nucleic Acids Res. 27(13): (1999) Aucun programme n’est meilleur que les autres La qualité de l’alignement dépend de l’ensemble de séquences (séquences similaires, divergentes, courtes, longues, ...) Il y a des erreurs systématiques lorsqu’on aligne des séquences dans la twilight zone (20-30% d’identité) Briffeuil P. et al. Bioinformatics 4: (1998) Le consensus de plusieurs méthodes augmente la sélectivité Lambert C. et al. Current Genomics 4: (2003) La combinaison de plusieurs méthodes peut augmenter la précision
: (1999) Aucun programme n’est meilleur que les autres. La qualité de l’alignement dépend de l’ensemble de séquences (séquences similaires, divergentes, courtes, longues, ...) Il y a des erreurs systématiques lorsqu’on aligne des séquences dans la twilight zone (20-30% d’identité) Briffeuil P. et al. Bioinformatics 4: (1998) Le consensus de plusieurs méthodes augmente la sélectivité. Lambert C. et al. Current Genomics 4: (2003) La combinaison de plusieurs méthodes peut augmenter la précision.")
30
Extraction des alignements pairés
ESyPAli Expert System for Pairwise Alignment Deux séquences PSI-BLAST Etape 1 Etape 2 Ensemble A Ensemble B PURGE Etape 3 Multalin T-COFFEE Dialign2 ClustalW Match-Box Extraction des alignements pairés

31
Extraction des alignements pairés
ESyPAli Extraction des alignements pairés Etape 4 Attribution d’un score aux positions alignées Extraction des positions alignées Fréquence

32
Attribution d’un score aux positions alignées
Séquence 1 L-G: 3 L-R: 2 L-D: 1 E-D: 4 E-E: 1 E-A: 1

33
Extraction des alignements pairés
ESyPAli Extraction des alignements pairés Etape 4 Attribution d’un score aux positions alignées Extraction des positions alignées Fréquence Choix de la position ayant le plus haut score comme point d’ancrage Elimination des positions incompatibles Points d’ancrage Construction de l’alignement consensus final Etape 5

34
Alignements incompatibles
1. A D L I I Y L R T S P E V A Y E 2. L P G T N I V L G A L P E D R H

35
Extraction des alignements pairés
ESyPAli Extraction des alignements pairés Etape 4 Attribution d’un score aux positions alignées Extraction des positions alignées Fréquence Choix de la position ayant le plus haut score comme point d’ancrage Elimination des positions incompatibles Points d’ancrage Construction de l’alignement consensus final Etape 5

36
ESyPAliNN Réseau neuronal Extraction des alignements pairés Etape 4
Attribution d’un score aux positions alignées Extraction des positions alignées Etape 5 Choix de la position ayant le plus haut score comme point d’ancrage Elimination des positions incompatibles Points d’ancrage Construction de l’alignement consensus final

37
Entraînement du réseau neuronal
Entrée Wik Entrée Sortie Sortie connue Yi Yk V V Séquence 1 Cachée Structure 1 L L ClustalW ClustalW I Match-Box Match-Box I Dialign2 Dialign2 I Multalin Multalin T PSI-BLAST PSI-BLAST L L T-COFFEE T-COFFEE

38
Utilisation du réseau neuronal
Entrée Cachée Sortie Y Y Séquence 1 Séquence 1 W W ClustalW ClustalW T Match-Box Match-Box T Dialign2 Dialign2 T Multalin Multalin Y PSI-BLAST PSI-BLAST W W T-COFFEE T-COFFEE

39
Résultats de l’évaluation sur 202 alignements pairés
% identité inférieur à 36 % %ID moyen = 20% Programme Sensibilité (%) Sélectivité (%) Alignement de structures ,0 100,0 MULTALIN ,5 44,0 MATCHBOX ,1 27,8 DIALIGN ,9 48,1 PSIBLAST ,2 47,8 CLUSTALW ,9 29,2 T-COFFEE ,5 47,4 ESyPAli ,2 36,8 ESyPAliNN ,8 38,9
 Sélectivité (%) Alignement de structures 100,0 100,0. MULTALIN 25,5 44,0. MATCHBOX 27,1 27,8. DIALIGN2 33,9 48,1. PSIBLAST 35,2 47,8. CLUSTALW 35,9 29,2. T-COFFEE 37,5 47,4. ESyPAli 44,2 36,8. ESyPAliNN 46,8 38,9.")
40
Alignement pairé Conclusions ESyPAli
Combinaison efficace d’alignements ESyPAliNN Utilisation de réseaux neuronaux Alignements de meilleure qualité

41
Plan Introduction Buts Développement de ESyPAliNN
Développement de ESyPred3D Base de données Brucella melitensis Conclusions / perspectives

42
ESyPAliNN Alignement Cible-template
ESyPred3D Expert System to Predict 3D structures of proteins C. Lambert et al., Bioinformatics, 18(9): (2002) Entrée Séquence cible PDB template PSI-BLAST, nr (NCBI) ESyPAliNN Alignement Cible-template MODELLER v6.2 Procheck Résultat Structure cible prédite
: (2002) Entrée. Séquence cible. PDB. template. PSI-BLAST, nr (NCBI) ESyPAliNN Alignement Cible-template. MODELLER v6.2. Procheck. Résultat. Structure cible prédite.")
43
Qualité de l’étape ESyPAli au concours CASP4
C. Lambert et al., Bioinformatics, 18(9): (2002)
: (2002)")
44
ESyPred3D EVA Système d’évaluation continue de serveurs de modélisation par homologie (catégorie CM) ESyPred3D , 3D-Jigsaw , Swiss-Model 853 modèles de janvier à juin 2003

45
ESyPred3D CASP5 CASP5 ESyPred3D parmi les 10 meilleurs serveurs évalués (sur 55) ESyPred3D dans le top 40 (si modélisateurs humains + serveurs (180)) Améliorer possible de la sélection du template
) Améliorer possible de la sélection du template.")
46
ESyPred3D Conclusions ESyPred3D est un des meilleurs serveurs de modélisation Performances dues essentiellement à ESyPAliNN

47
Plan Introduction Buts Développement de ESyPAli(NN)
Développement de ESyPred3D Base de données Brucella melitensis Conclusions / perspectives

48
Banque de données structurales
Intérêt Prédiction (détermination) de structures 3D à l’échelle d’un génome 1) Structure (prédite) accessible Ingénierie, hypothèses mutations 2) Recherche de sites actifs Aide à la détermination de fonction 3) Screening (docking) de petites molécules Recherche de cibles pour antibiotiques
 de structures 3D à l’échelle d’un génome. 1) Structure (prédite) accessible. Ingénierie, hypothèses mutations. 2) Recherche de sites actifs. Aide à la détermination de fonction. 3) Screening (docking) de petites molécules. Recherche de cibles pour antibiotiques.")
49
Banque de données Qualité des données
Problème définition de la position des codons start des pCDS de Integrated Genomics Inc. ----> Correction: consortium de spécialistes de Brucella Fonction prédite par similarité: BLAST/Swiss-Prot et hmmer/Pfam Localisation cellulaire: PSORT Prédiction des structures secondaires: PSI-PRED2 Application de ESyPred3D aux protéines déduites du génome de Brucella melitensis

50
Objectifs atteints de la banque de données
Centraliser les informations à propos du génome de Brucella Fournir des prédictions pour faciliter l’annotation et la rendre plus fiable Corriger les informations de la base de données suivant les modifications des utilisateurs Effectuer des recherches avancées Aider à coordonner des corrections à l’échelle génomique Intégrer des données biologiques pertinentes

51
Plan Introduction Buts Développement de ESyPAli(NN)
Développement de ESyPred3D Base de données Brucella melitensis Conclusions / perspectives

52
Conclusion Développement d’une méthode fiable d’alignement pairé de séquences Utilisation de ce nouveau programme dans une méthode automatique de modélisation par homologie Développement d’une banque de données structurales et fonctionnelles Développement d’ Utilisation de Coordtnation de

53
Perspectives Alignement de séquences
Développer une méthode d’alignement multiple Modélisation par homologie Amélioration de l’identification du template Base de données Docking de petites molécules Prédiction d’interactions protéine-protéine

54
Remerciements Eric Depiereux Benjamin Guy Baudoux URBM Monique
Nadia Johan Katalin et Bernard Nicolas et Isabelle Marc Marti-Renom (UCSF) Volker Eirich (Columbia) Ernest Feytmans (SIB) Benjamin URBM Aïko et Etienne Xavier Jean-Jacques Jean VDH Labo CMS Labo CTA Parents
 Volker Eirich (Columbia) Ernest Feytmans (SIB) Benjamin. URBM. Aïko et Etienne. Xavier. Jean-Jacques. Jean VDH. Labo CMS. Labo CTA. Parents.")
Présentations similaires




















 –Traitement difficile : 4 à 6 molécules, parmi lesquelles.>")