Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Identification et analyse bioinformatiques d'éléments cis-régulateurs
dans les génomes Bernard Jacq, M2 BBSG 2008 Module GF 1

2
Plan du cours (1ère partie) Rappels biologiques
Introduction Les éléments cis-régulateurs (ADN) Les éléments trans-régulateurs (protéines) Régions régulatrices et régulation in vivo 2
 Les éléments trans-régulateurs (protéines) Régions régulatrices et régulation in vivo. 2.")
3
LE CONTROLE DE L’INFORMATION GENETIQUE EUCARYOTE
TRANSCRIPTION TRADUCTION ADN ARN PROTEINE 3

4
Quelques généralités sur la régulation de la transcription eucaryote (1/2)
La variabilité d’expression des gènes eucaryotes résulte d’interactions entre des régions particulières des gènes et des combinaisons de facteurs protéiques spécifiques Ces régions d ’ADN sont d’une part les promoteurs (éléments génériques) et d’autres part différents types de régions appelées éléments cis-régulateurs (spécifiques) Le promoteur est défini comme la région d’ADN immédiatement en amont du site d’initiation de la transcription, au niveau de laquelle s’assemble le complexe d’initiation de la transcription (polymérase, cofacteurs) 4
 et d’autres part différents types de régions appelées éléments cis-régulateurs (spécifiques) Le promoteur est défini comme la région d’ADN immédiatement en amont du site d’initiation de la transcription, au niveau de laquelle s’assemble le complexe d’initiation de la transcription (polymérase, cofacteurs) 4.")
5
Quelques généralités sur la régulation de la transcription eucaryote (2/2)
Il y a différentes classes d’éléments cis-régulateurs: enhanceurs silenceurs insulateurs sites d’attachement à la matrice chromosomique • l’ensemble des éléments cis-régulateurs d’un gène a généralement une organisation modulaire: différents sous-éléments cis-régulateurs sont chacun responsables d’une partie du patron d’expression global du gène • les différents modules interagissent avec le même promoteur et la machinerie de transcription qui y est fixée 5

6
LE CONTROLE COMPLEXE DE L’ACTIVITE D’UN GENE PAR PLUSIEURS
INTERACTIONS PROTEINE-ADN 6

7
Plan du cours (1ère partie)
Introduction Les éléments cis-régulateurs (ADN) Les éléments trans-régulateurs (protéines) Régions régulatrices et régulation in vivo 2
 Les éléments trans-régulateurs (protéines) Régions régulatrices et régulation in vivo. 2.")
8
Les Promoteurs eucaryotes
Ils sont consistués d’un groupe de motifs d’ADN regroupés en une 100aine de pb en 5’ du site d’initiation TATA Box • INR Box DPE Box 7

9
Promoteur La TATA box est reconnue par la TBP (TATA binding Protein). L’ INR (initiator) et le DPE (downstream promoter element) sont reconnus par des TAFs (facteurs de transcription)
 et le DPE (downstream promoter element) sont reconnus par des TAFs (facteurs de transcription)")
10
Exemples de TATA Boxes 8

11
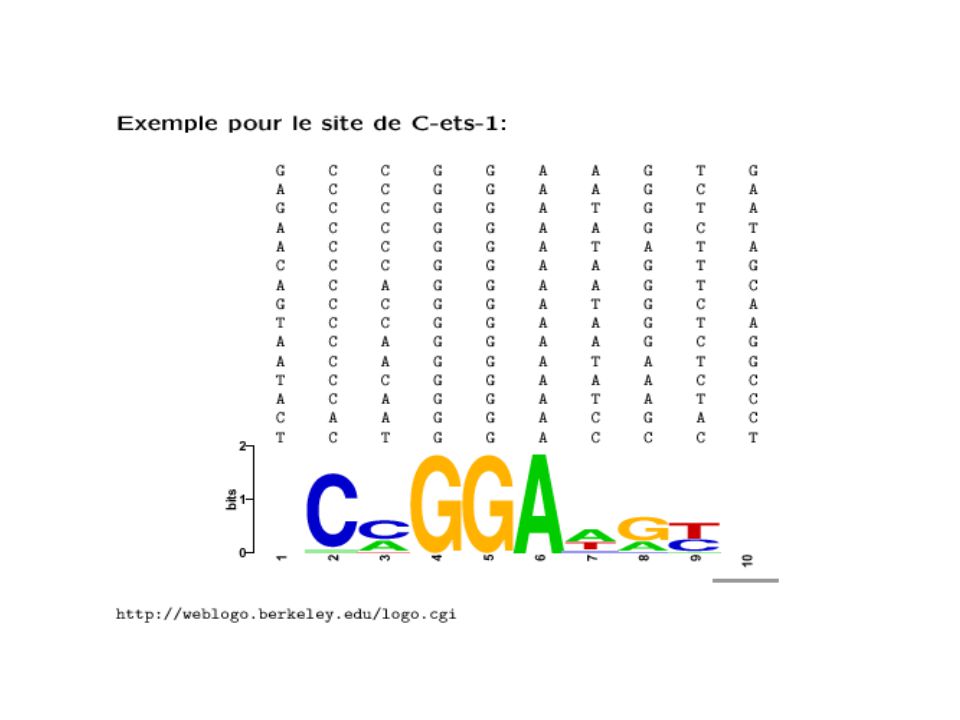
Une façon de présenter des séquences consensus:
Les « sequence logos » 9

12
Les Enhanceurs • Aussi appelées Upstream Activation Sequences (UAS)
• Séquences qui assistent le promoteur dans l’initiation • Peuvent être situées en amont ou en aval du promoteur (et même du gène) • Peuvent être actives dans l’une ou l’autre orientation • Sont reconnues par des facteurs de transcription spécifiques 11
 • Peuvent être actives dans l’une ou l’autre orientation. • Sont reconnues par des facteurs de transcription spécifiques. 11.")
13
Plan du cours (1ère partie)
Introduction Les éléments cis-régulateurs (ADN) Les éléments trans-régulateurs (protéines) Régions régulatrices et régulation in vivo 2
 Les éléments trans-régulateurs (protéines) Régions régulatrices et régulation in vivo. 2.")
14
Les Protéines régulatrices de la transcription chez les eucaryotes
• Les éléments cis-régulateurs de la transcription sont reconnus par un grand nombre de facteurs protéiques : • certains sont ubiquitaires : polymérase sur promoteur et FT généraux • la majorité sont spécifiques : facteurs de transcription sur les enhanceurs • Il existe plusieurs centaines de facteurs de transcription différents chez chaque organisme eucaryote, regroupés en quelques dizaines de familles structurales 14

15
Les principales familles de protéines régulatrices
Protéines à motifs HTH Protéines à doigts à zinc Protéines Leucine zipper 16

16
Les principales familles de protéines régulatrices
Protéines à motifs HTH Protéines à doigts à zinc Protéines Leucine zipper 16

17
Plan du cours (1ère partie)
Introduction Les éléments cis-régulateurs (ADN) Les éléments trans-régulateurs (protéines) Modules cis-régulateurs et régulation in vivo 2
 Les éléments trans-régulateurs (protéines) Modules cis-régulateurs et régulation in vivo. 2.")
18
Modules régulateurs Des gènes exprimés dans des contextes spatio-temporaux spécifiques possèdent souvent des MCR (modules cis-régulateurs) spécifiques : séquence régulatrice possédant un nombre significativement élevé de sites de fixation pour des facteurs de transcription tissu-spécifiques.
 spécifiques : séquence régulatrice possédant un nombre significativement élevé de sites de fixation pour des facteurs de transcription tissu-spécifiques.")
19
Beaucoup de genes eucaryotes sont controlés par des combinaisons d’activateurs et de répresseurs

20
Quelques exemples d’éléments cis-régulateurs chez la Drosophile
stripes 3 + 7 stripe 2 repression site cluster ps 6,8,10,12 stripes blastoderm + mesoderm at GBE Quelques exemples d’éléments cis-régulateurs chez la Drosophile ftz zebra element eve stripes 2 & 3+7 elements rho lateral neurectoderm stripe element kni posterior element Ubx PBX element 27

21
Plan du cours (2ème partie) Bioinformatique
Motifs de fixation à l’ADN Recherche de sites Découverte de sites Recherche de sites à l ’échelle génomique

22
Les problèmes spécifiques liés à la recherche de motifs d’interaction dans l’ADN
Les motifs sont courts (6 à 20 pb) Ils utilisent un alphabet limité (A,C,G,T) Ils peuvent présenter une variabilité de séquence importante (sont dégénérés) Ils possèdent un faible contenu d’information (8 à 12 bits, soit un site tous les pb) L’affinité de différents sites pour une même protéine peut varier de 3 ordres de magnitude La fonction régulatrice dépend souvent d ’interactions coopératives avec des sites voisins Ces sites sont trouvés dans les régions non-codantes Celles-ci représentent: - 11% du génome d’E. coli - 25% du génome de la levure - 97% du génome humain 63
 Ils utilisent un alphabet limité (A,C,G,T) Ils peuvent présenter une variabilité de séquence importante (sont dégénérés) Ils possèdent un faible contenu d’information (8 à 12 bits, soit un site tous les pb) L’affinité de différents sites pour une même protéine peut varier de 3 ordres de magnitude. La fonction régulatrice dépend souvent d ’interactions coopératives avec des sites voisins. Ces sites sont trouvés dans les régions non-codantes. Celles-ci représentent: - 11% du génome d’E. coli. - 25% du génome de la levure. - 97% du génome humain. 63.")
23
L’analyse bioinformatique de régions 2 situations différentes
cis-régulatrices 2 situations différentes 1) Recherche de motifs connus dans une ou plusieurs séquences - Expressions régulières - Matrices consensus 2) Découverte de motifs : Un groupe de séquences régulatrices doit contenir des motifs identiques ou similaires pour un ou plusieurs facteurs de transcription, mais ces motifs sont inconnus 31
 Recherche de motifs connus dans une ou plusieurs séquences. - Expressions régulières. - Matrices consensus. 2) Découverte de motifs : Un groupe de séquences régulatrices doit contenir des motifs identiques ou similaires pour un ou plusieurs facteurs de transcription, mais ces motifs sont inconnus. 31.")
24
Plan du cours (2ème partie)
Motifs de fixation à l’ADN Recherche de sites Découverte de sites Recherche de sites à l ’échelle génomique

25
Différentes représentations d'un motif -> recherche dans de nouvelles séquences
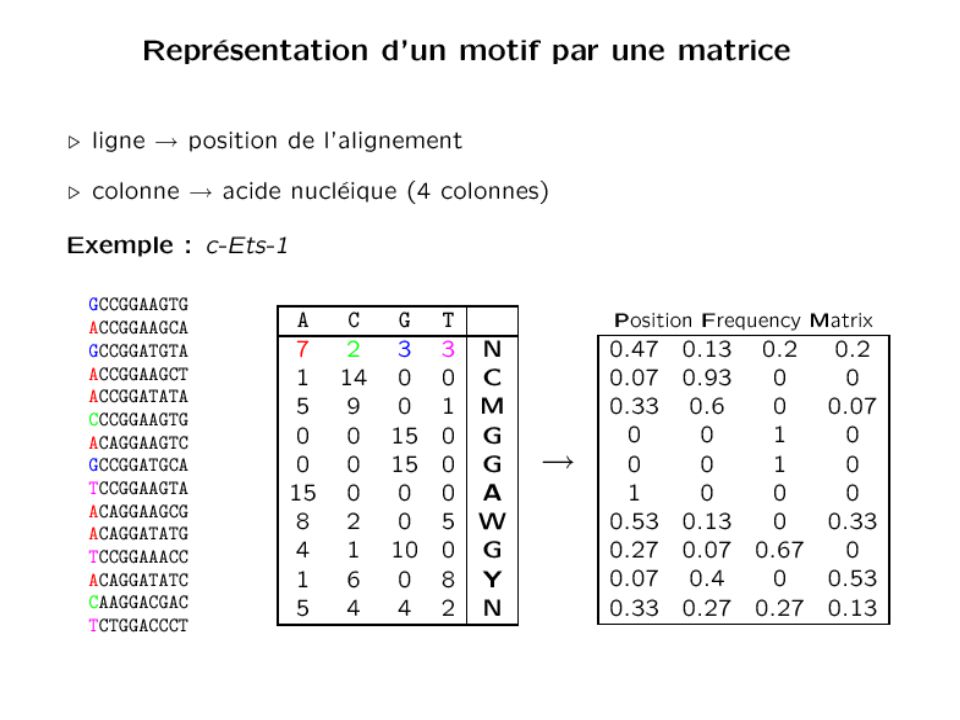
AAAAGAGTCA AAATGACTCA AAGTGAGTCA GGATGAGTCA AAATGAGTCA GAATGAGTCA YYYWGASTCA Collection de sites -> recherche d'expressions régulières (avec/sans substitution/délétion/insertion) Séquence consensus (utilisation du code IUPAC) -> recherche d'une expression régulière A T G C Matrice de fréquence + "scoring scheme" + seuil -> application lors du parcours de séquence Sequence logo Représentation du contenu informationnel (en bits) 33
 Séquence consensus (utilisation du code IUPAC) -> recherche d une expression régulière A T G C Matrice de fréquence + scoring scheme + seuil. -> application lors du parcours de séquence. Sequence logo. Représentation du contenu. informationnel (en bits) 33.")
28
Passage d ’une matrice de fréquence à une matrice de poids-position

29
Sites régulateurs: matrices pondérées
Site reconnu par le facteur Pho4p de la levure (Source : TRANSFAC) Valeur entre 0 et 2 (bits) seq(i) = fb,i log2 b fb,i pb Fréquence observée de chaque base à la position i Position Fréquence génomique de la base b Crédit: Denis Thieffry 36
 Valeur entre 0 et 2 (bits) seq(i) = fb,i log2. b. fb,i. pb. Fréquence observée de chaque base à la position i. Position. Fréquence génomique de la base b. Crédit: Denis Thieffry. 36.")
30
Recherche d'un motif avec une matrice pondérée
La séquence est parcourue avec la matrice, et un score est calculé pour chaque position Le plus haut score reflète la plus grande probabilité d'avoir un site fonctionel Comment définir un seuil significatif ? 38

31
Evaluation des outils de recherche de sites
Constitution de collections de séquences contrôles: + contenant des sites caractérisés - ne contenant assurément pas de sites - séquences "aléatoires" - séquences "brouillées" Différentes situations: Site correctement reconnu = "vrai positif" Prédiction abusive = "faux positif" Site manqué = "faux négatif" Absence de site correctement prédite = "vrai négatif" Crédit: Denis Thieffry 39

32
Compromis lors de la recherche de sites entre:
Grande sélectivité faible sensibilité -> grande confiance dans les sites prédits mais beaucoup de sites réels sont manqués Faible sélectivité grande sensibilité -> les sites réels sont noyés dans une mer de faux positifs => Double évaluation: Sélectivité = Nbre vrais positifs/ Nbre total hits Sensibilité = Nbre vrais positifs/ Nbre total sites Total sites = vrais positifs + faux négatifs Total "hits" = vrais positifs + faux positifs Crédit: Denis Thieffry 40

33
EVALUATION DES PERFORMANCES
VRAIS NEGATIFS ENSEMBLE DE SEQUENCES FAUX POSITIFS PROMOTEURS VRAIS PROMO TEURS TROU VES PROMOTEURS CORRECTE- MENT PREDITS VRAIS POSITIFS FAUX NEGATIFS COMMENT EVALUER LES PERFORMANCES DE LA METHODE ? TAUX DE RAPPEL = VRAIS POSITIFS/VRAIS POSITIFS +FAUX NEGATIFS TAUX DE PRECISION = VRAIS POSITIFS/VRAIS POSITIFS +FAUX POSITIFS NB: TAUX DE PRECISION ET DE RAPPEL VARIENT EN SENS INVERSE 41

34
Compromis lors de la recherche de sites
nombre de sites reconnus vrais négatifs vrais positifs faux négatifs faux positifs score seuil Crédit: Denis Thieffry 43

35
Pouvoir discriminant d'une matrice
Fortement discriminant Faiblement discriminant Faux positifs Vrai positifs Total hits Faux positifs Vrai positifs Total hits Fréquence Fréquence Score Score Raisonnablement discriminant Faux positifs Vrai positifs Total hits Fréquence Score Crédit: Denis Thieffry 44

37
Sites de facteurs de transcription de Drosophile

38
Pause …..

39
Plan du cours (2ème partie)
Plan (2ème partie) Plan du cours (2ème partie) Motifs de fixation à l’ADN Recherche de sites Découverte de sites Recherche de sites à l ’échelle génomique
 Plan du cours (2ème partie) Motifs de fixation à l’ADN. Recherche de sites. Découverte de sites. Recherche de sites à l ’échelle génomique.")
40
Algorithmes de découverte de motifs
Fréquences d'oligonucleotides (W)consensus Algorithmes stochastiques Gibbs sampling (AlignACE) Maximum expectation (MEME) HMM Neural networks 46
consensus. Algorithmes stochastiques. Gibbs sampling (AlignACE) Maximum expectation (MEME) HMM. Neural networks. 46.")
41
Découverte de motifs: Gibbs sampler (A Neuwalt)
Faites comme si vous connaissiez le motif, cela pourrait bien devenir vrai... 1) Sélection d'un nombre aléatoire de positions dans un ensemble de séquences 2) Création d'une matrice avec ces positions 3) Isolation d'une séquence de l'ensemble et recherche du meilleur score avec la matrice 4) Remplacement de l'ancienne position avec la nouvelle position et actualisation de la matrice 5) Recommencer à partir de (3) pour un nombre fixe de cycles Trouvé A C G T N itérations Pas trouvé Crédit: Denis Thieffry 48
 Sélection d un nombre aléatoire de positions dans un ensemble de séquences. 2) Création d une matrice avec ces positions. 3) Isolation d une séquence de l ensemble et recherche du meilleur score avec la matrice. 4) Remplacement de l ancienne position avec la nouvelle position et actualisation de la matrice. 5) Recommencer à partir de (3) pour un nombre fixe de cycles. Trouvé. A C G T N itérations. Pas trouvé. Crédit: Denis Thieffry. 48.")
42
Gibbs Sampling Example
The following slides illustrate Gibbs sampling to discover a motif in yeast DNA sequences. This example uses a sequence model that allows multiple sites per sequence. Columns are sampled as well as sites.

43
The Input Data Set 5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 bp of upstream sequence per gene are searched in Saccharomyces cerevisiae. Source: G.M. Church

44
The Target Motif (ce qu’il faudra découvrir)
5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 AAAAGAGTCA AAATGACTCA AAGTGAGTCA AAAAGAGTCA GGATGAGTCA AAATGAGTCA GAATGAGTCA AAAAGAGTCA MAP score = (maximum) ********** Source: G.M. Church
 ********** Source: G.M. Church.")
45
Initial Seeding MAP score = -10.0 Source: G.M. Church
5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT 5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 TGAAAAATTC TGAAAAATTC GACATCGAAA GACATCGAAA GCACTTCGGC GCACTTCGGC GAGTCATTAC GAGTCATTAC GTAAATTGTC GTAAATTGTC CCACAGTCCG CCACAGTCCG TGTGAAGCAC TGTGAAGCAC MAP score = -10.0 ********** ********** Source: G.M. Church

46
How much better is the alignment with this site as opposed to without?
Sampling Add? 5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 TCTCTCTCCA TGAAAAATTC How much better is the alignment with this site as opposed to without? TGAAAAATTC GACATCGAAA GACATCGAAA GCACTTCGGC GCACTTCGGC GAGTCATTAC GAGTCATTAC GTAAATTGTC GTAAATTGTC CCACAGTCCG CCACAGTCCG TGTGAAGCAC TGTGAAGCAC ********** ********** Source: G.M. Church

47
How much better is the alignment with this site as opposed to without?
Continued Sampling Add? Remove. 5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 ATGAAAAAAT TGAAAAATTC How much better is the alignment with this site as opposed to without? TGAAAAATTC GACATCGAAA GACATCGAAA GCACTTCGGC GCACTTCGGC GAGTCATTAC GAGTCATTAC GTAAATTGTC GTAAATTGTC CCACAGTCCG CCACAGTCCG TGTGAAGCAC TGTGAAGCAC ********** ********** Source: G.M. Church

48
How much better is the alignment with this site as opposed to without?
Continued Sampling Add? 5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 How much better is the alignment with this site as opposed to without? TGAAAAATTC GACATCGAAA GACATCGAAA GCACTTCGGC GCACTTCGGC GAGTCATTAC GAGTCATTAC GTAAATTGTC GTAAATTGTC CCACAGTCCG CCACAGTCCG TGTGAAGCAC TGTGAAGCAC ********** ********** Source: G.M. Church

49
How much better is the alignment with this new column structure?
Column Sampling 5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 How much better is the alignment with this new column structure? GACATCGAAA GACATCGAAAC GCACTTCGGC GCACTTCGGCG GAGTCATTAC GAGTCATTACA GTAAATTGTC GTAAATTGTCA CCACAGTCCG CCACAGTCCGC TGTGAAGCAC TGTGAAGCACA ********** ********* * Source: G.M. Church

50
The Best Motif MAP score = 20.37 Source: G.M. Church
5’- TCTCTCTCCACGGCTAATTAGGTGATCATGAAAAAATGAAAAATTCATGAGAAAAGAGTCAGACATCGAAACATACAT …HIS7 5’- ATGGCAGAATCACTTTAAAACGTGGCCCCACCCGCTGCACCCTGTGCATTTTGTACGTTACTGCGAAATGACTCAACG …ARO4 5’- CACATCCAACGAATCACCTCACCGTTATCGTGACTCACTTTCTTTCGCATCGCCGAAGTGCCATAAAAAATATTTTTT …ILV6 5’- TGCGAACAAAAGAGTCATTACAACGAGGAAATAGAAGAAAATGAAAAATTTTCGACAAAATGTATAGTCATTTCTATC …THR4 5’- ACAAAGGTACCTTCCTGGCCAATCTCACAGATTTAATATAGTAAATTGTCATGCATATGACTCATCCCGAACATGAAA …ARO1 5’- ATTGATTGACTCATTTTCCTCTGACTACTACCAGTTCAAAATGTTAGAGAAAAATAGAAAAGCAGAAAAAATAAATAA …HOM2 5’- GGCGCCACAGTCCGCGTTTGGTTATCCGGCTGACTCATTCTGACTCTTTTTTGGAAAGTGTGGCATGTGCTTCACACA …PRO3 AAAAGAGTCA AAATGACTCA AAGTGAGTCA AAAAGAGTCA GGATGAGTCA AAATGAGTCA GAATGAGTCA AAAAGAGTCA MAP score = 20.37 ********** Source: G.M. Church

51
Plan du cours (2ème partie)
Motifs de fixation à l’ADN Recherche de sites Découverte de sites Recherche de sites à l’échelle génomique

52
Faisabilité d’une recherche de motifs sur l’ensemble d’un génome
Genome de la levure: (12,5 Mb) Sites de contrôle de la transcription (~7 bases d’information) 7 bases d’information (14 bits) ~ 1 occurence toutes les bases. Une moyenne de 1500 occurences dans un génome de 12 Mb (24 * 106 sites). Le nombre réel de sites biologiquement significatifs est probablement beaucoup plus faible . 75
 Sites de contrôle de la transcription. (~7 bases d’information) 7 bases d’information (14 bits) ~ 1 occurence toutes les bases. Une moyenne de 1500 occurences dans un génome de 12 Mb. (24 * 106 sites). Le nombre réel de sites biologiquement significatifs est probablement beaucoup plus faible")
53
On peut mettre à profit :
Comment réduire l’espace de recherche dans les séquences pour une recherche génomique ? (1) On peut mettre à profit : les données d’expression d’ARN (microarrays): clusterisation en utilisant les données de mutations, les conditions expérimentales, les cinétiques d’expression. les données d’expression spatio-temporelle: hybridations in situ chez les métazoaires les catégorisations fonctionnelles des gènes (f. biochimique, f. cellulaire) 76
 On peut mettre à profit : les données d’expression d’ARN (microarrays): clusterisation en utilisant les données de mutations, les conditions expérimentales, les cinétiques d’expression. les données d’expression spatio-temporelle: hybridations in situ chez les métazoaires. les catégorisations fonctionnelles des gènes (f. biochimique, f. cellulaire) 76.")
54
On peut mettre à profit :
Comment réduire l’espace de recherche dans les séquences pour une recherche génomique ? (2) On peut mettre à profit : la conservation inter-spécifique (phylogenomic footprinting) la sélection des séquences: éliminer les régions codantes (ORFs), les régions répétitives, et toute séquence susceptible de ne pas contenir de régions de régulation Le regroupement (clusterisation) de sites identiques: il est rare de trouver un seul site pour un facteur de transcription donné dans une région cis-régulatrice confirmée) ou de sites pour des FT de même spécificité Spatio-temporelle 76
 On peut mettre à profit : la conservation inter-spécifique (phylogenomic footprinting) la sélection des séquences: éliminer les régions codantes (ORFs), les régions répétitives, et toute séquence susceptible de ne pas contenir de régions de régulation. Le regroupement (clusterisation) de sites identiques: il est rare de trouver un seul site pour un facteur de transcription donné dans une région cis-régulatrice confirmée) ou de sites pour des FT de même spécificité Spatio-temporelle. 76.")
55
Des Microarrays à la découverte de motifs cis-régulateurs
EMBL start Clustering A1234 Z4321 Microarrays Blast start Recherche de motifs D ’après Magali Lescot

56
Comparaisons Homme-souris genome pour localiser des sites de régulation (Phylogenomic footprinting)
"98% of experimentally defined ... binding sites of skeletal-muscle-specific transcription factors are confined to the 19% of human sequences that are most conserved in the orthologous rodent sequences ... the binding specificities of all three major ... factors (MYF, SRD & MEF2) can be computationally identified." Wasserman et al, Nat Genet 2000 Oct;26(2):225-8 80
 can be computationally identified. Wasserman et al, Nat Genet 2000 Oct;26(2):")
57
Phylogenomic footprinting: utilisation du programme Vista

58
Exemple d ’utilisation de la notion de clusters
de sites différents

59
Exemple d ’utilisation de la notion de clusters
de sites différents

60
Exemple d ’utilisation de la notion de clusters
de sites différents

61
(M. Caselle, Université de Turin)
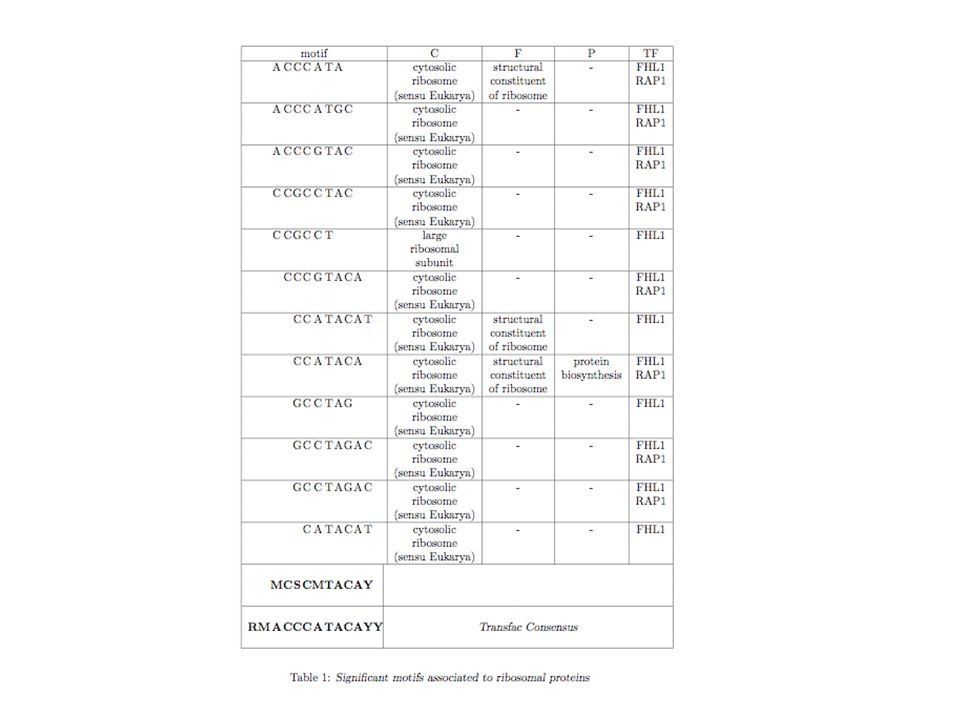
Une autre aproche (M. Caselle, Université de Turin) Our Approach. first step Grouping of genes based on the motifs that are overrepresented in their upstream regions. To each possible word w we associate the set Sw of all the genes in whose upstream region the word w is overrepresented second step Select those sets which show some kind of functional characterization using microarray experiments or Gene Ontology annotations. Microarray: For each set Sw we compare the expression distribution within the set with the genome wide one (using for example Kolmogorv- Smirnov test).
 Our Approach. first step Grouping of genes based on the motifs that are overrepresented in their upstream regions. To each possible word w we associate the set Sw of all the genes in whose upstream region the word w is overrepresented. second step Select those sets which show some kind of functional characterization using microarray experiments or Gene Ontology annotations. Microarray: For each set Sw we compare the expression distribution within the set with the genome wide one (using for example Kolmogorv- Smirnov test).")
63
Les sets S (mots) • Pour chaque mot (5 à 8 pb), calculer sa fréquence dans toutes les régions en 5’ des gènes de tout le génome considéré comme un échantillon unique. • On compte ensuite l’occurrence du mot dans la région 5’ de chaque gène pris isolément • Si le nombre d’occurrences du mot dans la région 5’ du gène G est statistiquement significative (comparé par exemple à une distribution binomiale basée sur les fréquences de référence ci-dessus, alors le gène G appartient au sous-ensemble S
, calculer sa fréquence dans toutes les régions en 5’ des gènes de tout le génome considéré comme un échantillon unique. • On compte ensuite l’occurrence du mot dans la région 5’ de chaque gène pris isolément. • Si le nombre d’occurrences du mot dans la région 5’ du gène G est statistiquement significative (comparé par exemple à une distribution binomiale basée sur les fréquences de référence ci-dessus, alors le gène G appartient au sous-ensemble S.")
64
Le Filtre GO (Gene Ontology)
Pour chaque sous-ensemble S, on calcule la prévalence de tous les termes GO parmi les gènes annotés de S et la probabilité qu’une telle prévalence puisse survenir au hasard dans un jeu de même taille de gènes choisis au hasard. On considère qu’un gène est annoté pour un terme GO t s’il est directement annoté avec ce terme ou l’un quelconque de ses descendants dans le graphe GO.

65
Pour un terme GO donné t, on appelle K(t) est le nombre total d’ORFs annotés avec ce terme dans le génome et K(m,t) le nombre d’ORFs annotés avec lui dans le set S(m). Si J et j(m) dénotent le nombre d’ORFs dans le génome et dans ke set S(m) respectivement, la probabilité du terme t est décrite par une loi hypergéométrique De cette façon, une p-value peut ête associée à chaque paire constituée d’un motif et d’un terme GO donnés Où

66
Taux de fausses découvertes
Le problème: vu le très grand nombre de P-values qui seront calculées (égal en principe au nombre de termes GO multiplié par le nombre de motifs analysés), il est clair que quelques valeurs faibles de P-values (faux positifs) peuvent apparaître dûes au hasard La façon classique de gérer ce problème (la correction de Bonferroni) n’est pas appropriée gans notre cas: A cause de la nature hiérarchique de l’ontologie GO, les différentes P-values calculées ne peuvent être considérées comme indépendantes les unes des autres.
, il est clair que quelques valeurs faibles de P-values (faux positifs) peuvent apparaître dûes au hasard. La façon classique de gérer ce problème (la correction de Bonferroni) n’est pas appropriée gans notre cas: A cause de la nature hiérarchique de l’ontologie GO, les différentes P-values calculées ne peuvent être considérées comme indépendantes les unes des autres.")
67
Comment gérer ce problème ?
Les auteurs proposent de générer un grand nombre Nr de sets de gènes comparable en taille à la taille typique des sets associés aux motifs puis de classer ces sets « random » sur la base de leurs meilleures P-values De cette façon, on peut déterminer une probabilité p f(C) de taux de fausse découverte qui soit une fonction du seuil des P-values C
 de taux de fausse découverte qui soit une fonction du seuil des P-values C.")
75
Quelques problèmes d’actualité en bioinformatique
des séquences régulatrices Peut-on prédire la localisation de sites de fixation de FT sur une séquence ? Peut-on prédire la localisation d’un (ou plusieurs) promoteurs sur une séquence ? Peut-on prédire l’emplacement de régions régulatrices sur une séquence ? Echelle de facilité de la tâche Assez Facile Difficile 28
 promoteurs sur une séquence Peut-on prédire l’emplacement de régions régulatrices sur une séquence Echelle de facilité de la tâche. Assez Facile Difficile. 28.")
76
Quelques problèmes d’actualité en bioinformatique
des séquences régulatrices (suite) Peut-on comparer des régions régulatrices ? Peut-on prédire parmi des sites pour des FT ceux qui sont vraisemblables biologiquement et ceux qui ne le sont pas ? Peut-on prédire l’expression spatiale d’un gène par l’analyse de ses régions régulatrices ? 29
 Peut-on comparer des régions régulatrices Peut-on prédire parmi des sites pour des FT ceux qui sont vraisemblables biologiquement et ceux qui ne le sont pas Peut-on prédire l’expression spatiale d’un gène par l’analyse de ses régions régulatrices 29.")
77
C’est fini ! 82

Présentations similaires







>")














>")


