Télécharger la présentation
La présentation est en train de télécharger. S'il vous plaît, attendez

1
Apparentement et consanguinité dans le panel 1000 Génomes
Mourad Sahbatou Fondation Jean DAUSSET – CEPH, Paris 8èmes Assises de Génétique Humaine et Médicale Lyon, 3-5 février 2016

2
Programme international commencé en 2008.
Programme international commencé en 2008. Phase finale (phase3) est disponible depuis nov Catalogue de variants génétiques sur 26 populations, individus indépendants, regroupées en 5 régions: Afrique, Europe, Asie de l’Est, Asie du Sud et Amériques. Source unique de données de séquençage du génome humain. Outil pour la génétique médicale et la génétique des populations. => Importance de bien caractériser ce panel de référence
 est disponible depuis nov Catalogue de variants génétiques sur 26 populations, 2504 individus indépendants, regroupées en 5 régions: Afrique, Europe, Asie de l’Est, Asie du Sud et Amériques. Source unique de données de séquençage du génome humain. Outil pour la génétique médicale et la génétique des populations. => Importance de bien caractériser ce panel de référence.")
3
Objectifs Détecter l’apparentement et la consanguinité dans le panel 1000 Génomes : Présence de consanguinité déjà notée dans le panel HapMap3 (Gazal, Sahbatou et al, 2014); L’apparentement a seulement été vérifié sur des informations généalogiques. Répondre à un problème méthodologique : Robustesse de ces estimations en présence de populations ayant des individus métissés,« admixed ».
; L’apparentement a seulement été vérifié sur des informations généalogiques. Répondre à un problème méthodologique : Robustesse de ces estimations en présence de populations ayant des individus métissés,« admixed ».")
4
f coefficient de consanguinité génomique
Conséquence génétique de la consanguinité : - allèles identiques hérités d’un ancêtre commun (IBD) - les génotypes sont homozygotes par descendance (HBD) Enfant de cousin germain (1C) Segments HBD sur le génome Coefficient génomique f : proportion du génome HBD Différentes méthodes pour estimer f en utilisant des marqueurs génétiques Méthodes Simple Point Méthodes Multipoints Marqueur par marqueur Plusieurs marqueurs adjacents PLINK (Purcell et al. 2007) FSuite (Leutenegger et al ; Gazal et al. 2014)
 - les génotypes sont homozygotes par descendance (HBD) Enfant de cousin germain (1C) Segments HBD sur le génome. Coefficient génomique f : proportion du génome HBD. Différentes méthodes pour estimer f en utilisant des marqueurs génétiques. Méthodes Simple Point. Méthodes Multipoints. Marqueur par marqueur. Plusieurs marqueurs adjacents. PLINK (Purcell et al. 2007) FSuite (Leutenegger et al ; Gazal et al. 2014)")
5
FSuite Sans connaitre la généalogie:
- Détecter et estimer f par individu - Inférer le type d’apparentement entre les parents d’un individu 1C; 2C; 2x1C; AV. - Estimer la proportion de ce type de mariage dans une population 1C: cousins germains; 2C: cousins de 2nd degré 2x1C: double cousins germains; AV: oncle/nièce;

6
Estimation de f par FSuite
Dépend des fréquences alléliques Génotypes observés Modèle de Markov caché (HMM) HBD non observés Dépend des paramètres du modèle (f et a) et des distances génétiques Paramètres du modèle : f est la probabilité d’être HBD à un marqueur Les longueurs des segments HBD ont une distribution exponentielle de taille moyenne 1/a Estimation : f et a par maximum de vraisemblance Hypothèse d’une population homogène : dans laquelle on peut utiliser les même fréquences alléliques pour tous les individus dans le panel 1000 Génomes, cette hypothèse n’est plus respectée
 HBD non observés. Dépend des paramètres du modèle (f et a) et des distances génétiques. Paramètres du modèle : f est la probabilité d’être HBD à un marqueur. Les longueurs des segments HBD ont une distribution exponentielle de taille moyenne 1/a. Estimation : f et a par maximum de vraisemblance. Hypothèse d’une population homogène : dans laquelle on peut utiliser les même fréquences alléliques pour tous les individus. dans le panel 1000 Génomes, cette hypothèse n’est plus respectée.")
7
En présence de populations métissées
Les méthodes simple-point sont non valides. (Thornton et al. 2012, Moltke and Albrechtsen 2013) Qu’en est-il des méthodes multipoints telles que FSuite? Evaluation par simulation
 Qu’en est-il des méthodes multipoints telles que FSuite Evaluation par simulation.")
8
Résultats des simulations
100 réplicats d ’enfants de cousins germains (1C) Fréquences alléliques ADMCEU : Proportion du génome d’origine européenne (CEU) pour chaque individu simulé (ADMCEU = 0.8 => 0.2 proportion de génome africain) Fréquences européennes (CEU) : f valide pour les individus avec ADMCEU > 0.5 Fréquences africaines (YRI) : f valide pour les individus avec ADMCEU < 0.5 Fréquences asiatiques (JPT/CHB) : f non valide PLINK, méthode simple-point, f non valide quelles que soient les fréquences alléliques utilisées. Δf = festimé - fvrai
 Fréquences. alléliques. ADMCEU : Proportion du génome d’origine européenne (CEU) pour chaque individu simulé (ADMCEU = 0.8 => 0.2 proportion de génome africain) Fréquences européennes (CEU) : f valide pour les individus avec ADMCEU > 0.5. Fréquences africaines (YRI) : f valide pour les individus avec ADMCEU < 0.5. Fréquences asiatiques (JPT/CHB) : f non valide. PLINK, méthode simple-point, f non valide quelles que soient les fréquences alléliques utilisées. Δf = festimé - fvrai.")
9
Résultats des simulations
100 réplicats d ’enfants de cousins germains (1C) Fréquences alléliques ADMCEU : Proportion du génome d’origine européenne (CEU) pour chaque individu simulé (ADMCEU = 0.8 => 0.2 proportion de génome africain) Fréquences européennes (CEU) : f valide pour les individus avec ADMCEU > 0.5 Fréquences africaines (YRI) : f valide pour les individus avec ADMCEU < 0.5 Fréquences asiatiques (JPT/CHB) : f non valide Fréquences de l’échantillon (SAMPLE) : Δf = 0 f valide pour tout ADMCEU PLINK, méthode simple-point, f non valide lorsqu’on s’éloigne de ADMCEU = 0.5 . Δf = festimé - fvrai FSuite fournit des estimations fiables de f même en présence de personnes métissées dans la population quel que soit le degré de consanguinité
 Fréquences. alléliques. ADMCEU : Proportion du génome d’origine européenne (CEU) pour chaque individu simulé (ADMCEU = 0.8 => 0.2 proportion de génome africain) Fréquences européennes (CEU) : f valide pour les individus avec ADMCEU > 0.5. Fréquences africaines (YRI) : f valide pour les individus avec ADMCEU < 0.5. Fréquences asiatiques (JPT/CHB) : f non valide. Fréquences de l’échantillon (SAMPLE) : Δf = 0. f valide pour tout ADMCEU. PLINK, méthode simple-point, f non valide lorsqu’on s’éloigne de ADMCEU = Δf = festimé - fvrai. FSuite fournit des estimations fiables de f. même en présence de personnes métissées. dans la population quel que soit le degré de consanguinité.")
10
Application au panel 1000 Génomes
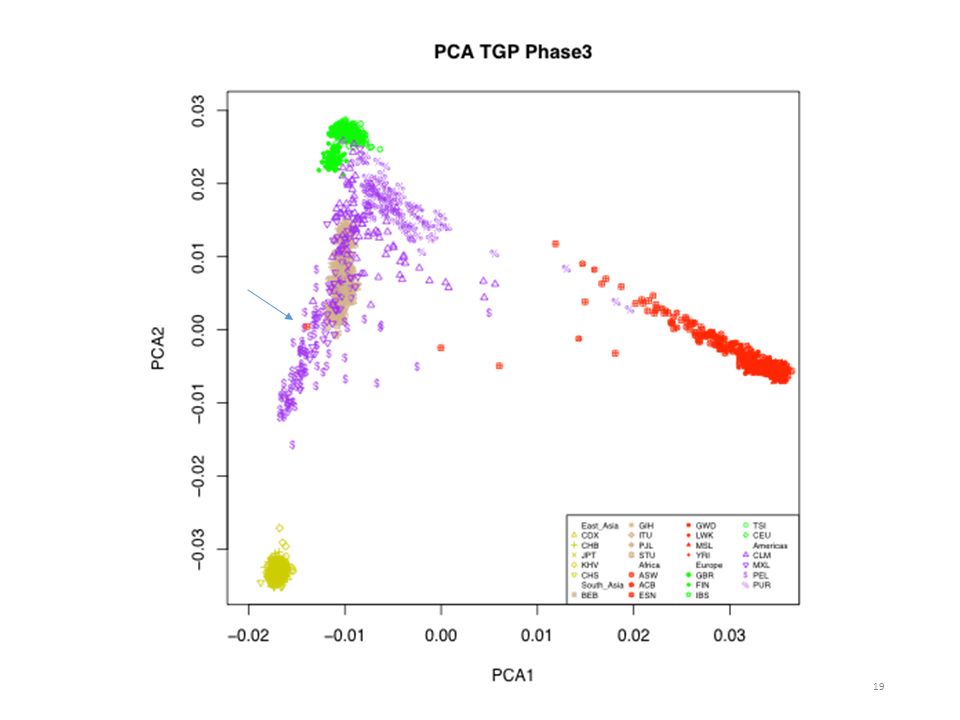
Final Phase African (AFR,7) 661 African Caribbean in Barbados (ACB) 96 African Ancestry in Southwest United States (ASW) 61 Esan in Nigeria (ESN) 99 Gambian in Western Division, The Gambia (GWD) 113 Luhya in Webuye, Kenya (LWK) Mende in Sierra Leone (MSL) 85 Yoruba in Ibadan, Nigeria (YRI) 108 European (EUR,5) 503 Utah residents with European ancestry (CEU) Finnish in Finland (FIN) British in England and Scotland (GBR) 91 Iberian populations in Spain (IBS) 107 Toscani in Italy (TSI) East Asian (EAS,5) 504 Chinese Dai in Xishuangbanna, China (CDX) 93 Han Chinese in Bejing, China (CHB) 103 Southern Han Chinese, China (CHS) 105 Japanese in Tokyo, Japan (JPT) 104 Kinh in Ho Chi Minh City, Vietnam (KHV) South Asian (SAS,5) 489 Bengali in Bangladesh (BEB) 86 Gujarati Indian in Houston,Texas (GIH) Indian Telugu in the United Kingdom (ITU) 102 Punjabi in Lahore, Pakistan (PJL) Sri Lankan Tamil in the United Kingdom (STU) Admixed American (ADM,4) 347 Colombian in Medellin, Colombia (CLM) 94 Mexican Ancestry in Los Angeles, California (MXL) 64 Peruvian in Lima, Peru (PEL) Puerto Rican in Puerto Rico (PUR) TOTAL 2,504 Application au panel 1000 Génomes 2 504 individus de 26 populations (5 régions) Présence de populations métissées FSuite est utilisé sur chaque population Calcul des fréquences alléliques par population (Freq. SAMPLE) Filtrage sur les polymorphismes fréquents (MAF>5%) : 81M -> 3M Variants
 661. African Caribbean in Barbados (ACB) 96. African Ancestry in Southwest United States (ASW) 61. Esan in Nigeria (ESN) 99. Gambian in Western Division, The Gambia (GWD) 113. Luhya in Webuye, Kenya (LWK) Mende in Sierra Leone (MSL) 85. Yoruba in Ibadan, Nigeria (YRI) 108. European (EUR,5) 503. Utah residents with European ancestry (CEU) Finnish in Finland (FIN) British in England and Scotland (GBR) 91. Iberian populations in Spain (IBS) 107. Toscani in Italy (TSI) East Asian (EAS,5) 504. Chinese Dai in Xishuangbanna, China (CDX) 93. Han Chinese in Bejing, China (CHB) 103. Southern Han Chinese, China (CHS) 105. Japanese in Tokyo, Japan (JPT) 104. Kinh in Ho Chi Minh City, Vietnam (KHV) South Asian (SAS,5) 489. Bengali in Bangladesh (BEB) 86. Gujarati Indian in Houston,Texas (GIH) Indian Telugu in the United Kingdom (ITU) 102. Punjabi in Lahore, Pakistan (PJL) Sri Lankan Tamil in the United Kingdom (STU) Admixed American (ADM,4) 347. Colombian in Medellin, Colombia (CLM) 94. Mexican Ancestry in Los Angeles, California (MXL) 64. Peruvian in Lima, Peru (PEL) Puerto Rican in Puerto Rico (PUR) TOTAL. 2,504. Application au panel 1000 Génomes individus de 26 populations (5 régions) Présence de populations métissées. FSuite est utilisé sur chaque population. Calcul des fréquences alléliques par population (Freq. SAMPLE) Filtrage sur les polymorphismes fréquents (MAF>5%) : 81M -> 3M Variants.")
11
Apparentement génomique
dans le panel 1000 génomes Tester l’apparentement de toutes les paires d’individus dans chaque population par RELPAIR (Epstein et al. 2000). 227 paires d’individus sont détectées comme apparentées Origine des apparentés 11 paires au 1er degré : 8 paires de parents-enfants dont 1 trio 3 paires de frères 4 paires au 2nd degré : 1 paire de demi-frère 3 paires de type oncle-nièce 212 paires de cousins germains 122 paires d’Afrique (AFR) 47 d’Asie du Sud (SAS) 22 d’Asie de l’Est (EAS) 21 des Amériques (AMR) 15 d’Europe (EUR)
. 227 paires d’individus sont détectées comme apparentées. Origine. des apparentés. 11 paires au 1er degré : 8 paires de parents-enfants dont 1 trio. 3 paires de frères. 4 paires au 2nd degré : 1 paire de demi-frère. 3 paires de type oncle-nièce. 212 paires de cousins germains. 122 paires d’Afrique (AFR) 47 d’Asie du Sud (SAS) 22 d’Asie de l’Est (EAS) 21 des Amériques (AMR) 15 d’Europe (EUR)")
12
Consanguinité génomique dans le panel 1000 Génomes
* * * * * * * * * * * 1/16 = 1/16ème du génome homozygote par descendance (6,25% HBD) 2x1C: double cousins germains; AV: oncle/nièce; 1C: cousins germains; 2C: cousins 2nd degré 595 individus sont inférés consanguins (24% du panel), essentiellement en Asie du Sud (SAS, 45%) ou en Amérique (AMR, 41%). Présence de consanguinité dans toutes les populations à des fréquences différentes : plus de 25% d’individus consanguins dans 11 populations; moins de 5% d’individus consanguins dans 6 populations; *
 2x1C: double cousins germains; AV: oncle/nièce; 1C: cousins germains; 2C: cousins 2nd degré. 595 individus sont inférés consanguins (24% du panel), essentiellement en Asie du Sud (SAS, 45%) ou en Amérique (AMR, 41%). Présence de consanguinité dans toutes les populations. à des fréquences différentes : plus de 25% d’individus consanguins dans 11 populations; moins de 5% d’individus consanguins dans 6 populations; *")
13
Consanguinité génomique dans le panel 1000 Génomes
1/16 = 1/16ème du génome homozygote par descendance (6,25% HBD) 2x1C: double cousins germains; AV: oncle/nièce; 1C: cousins germains; 2C: cousins 2nd degré Consanguinité éloignée : 501 individus inférés comme issus de couples de cousins au 2nd degré (2C) Consanguinité proche: 94 individus inférés comme issus de couples de cousins germains (1C) ou plus proche La population finlandaise (FIN), a un tiers des individus détectés consanguins (34%, tous 2C). Ceci est en accord avec l'histoire de la population finlandaise : un petit nombre de fondateurs et très peu d’immigration. Deux-tiers de ces individus consanguins (64/94) proviennent de 3 populations d’Asie du Sud : ITU (10), PJL (22), STU (32).
 2x1C: double cousins germains; AV: oncle/nièce; 1C: cousins germains; 2C: cousins 2nd degré. Consanguinité éloignée : 501 individus inférés comme issus de couples de cousins au 2nd degré (2C) Consanguinité proche: 94 individus inférés comme issus de couples de cousins germains (1C) ou plus proche. La population finlandaise (FIN), a un tiers des individus détectés consanguins (34%, tous 2C). Ceci est en accord avec l histoire de la population finlandaise : un petit nombre de fondateurs et très peu d’immigration. Deux-tiers de ces individus consanguins (64/94) proviennent de 3 populations d’Asie du Sud : ITU (10), PJL (22), STU (32).")
14
Panel sans apparentement, ni consanguinité très proche
TGP2457 Panel sans apparentement, ni consanguinité très proche (1er et 2nd degrés) TGP2261 Panel sans apparentement, ni consanguinité proche (1er, 2nd et 3ème degrés) supplément Table S4 (liste des 2504 individus)
 TGP2261. Panel sans apparentement, ni consanguinité proche. (1er, 2nd et 3ème degrés) supplément Table S4. (liste des 2504 individus)")
15
Conclusion Nous avons montré que les méthodes multipoints (Fsuite) fournissent des estimations fiables de f même en présence de populations métissées, contrairement aux méthodes simple-point (Plink). FSuite ( L’application aux données de séquençage de la phase finale du 1000 Génomes révèle la présence d’une forte proportion d’individus consanguins, essentiellement dans les populations d’Asie du Sud. 2 panels TGP2457 et TGP2261 sans consanguinité ni apparentement.
 fournissent des estimations fiables de f même en présence de populations métissées, contrairement aux méthodes simple-point (Plink). FSuite ( L’application aux données de séquençage de la phase finale. du 1000 Génomes révèle la présence d’une forte proportion. d’individus consanguins, essentiellement dans les populations. d’Asie du Sud. 2 panels TGP2457 et TGP2261 sans consanguinité. ni apparentement.")
16
Remerciements Steven Gazal, INSERM, APHP, Paris
Marie-Claude Babron, INSERM U946, Paris Emmanuelle Génin, INSERM U1078, Brest Anne-Louise Leutenegger, INSERM U946, Paris

18
Final Phase Phase 1 Pilot HapMap3 (9 pop/11) African (AFR) 661 246 208 231 African Caribbean in Barbados (ACB) 96 - African Ancestry in Southwest United States (ASW) 61 47 Esan in Nigeria (ESN) 99 Gambian in Western Division, The Gambia (GWD) 113 Luhya in Webuye, Kenya (LWK) 97 102 84 Mende in Sierra Leone (MSL) 85 Yoruba in Ibadan, Nigeria (YRI) 108 88 106 100 European (EUR) 503 379 160 193 Utah residents with European ancestry (CEU) 94 Finnish in Finland (FIN) 93 British in England and Scotland (GBR) 91 89 Iberian populations in Spain (IBS) 107 14 Toscani in Italy (TSI) 98 66 East Asian (EAS) 504 286 185 175 Chinese Dai in Xishuangbanna, China (CDX) Han Chinese in Bejing, China (CHB) 103 Southern Han Chinese, China (CHS) 105 Japanese in Tokyo, Japan (JPT) 104 90 Kinh in Ho Chi Minh City, Vietnam (KHV) South Asian (SAS) 489 Bengali in Bangladesh (BEB) 86 Gujarati Indian in Houston,Texas (GIH) Indian Telugu in the United Kingdom (ITU) Punjabi in Lahore, Pakistan (PJL) Sri Lankan Tamil in the United Kingdom (STU) Admixed American (ADM) 347 181 57 Colombian in Medellin, Colombia (CLM) 60 Mexican Ancestry in Los Angeles, California (MXL) 64 Peruvian in Lima, Peru (PEL) Puerto Rican in Puerto Rico (PUR) 55 TOTAL 2504 1092 553 747
 Esan in Nigeria (ESN) 99. Gambian in Western Division, The Gambia (GWD) 113. Luhya in Webuye, Kenya (LWK) Mende in Sierra Leone (MSL) 85. Yoruba in Ibadan, Nigeria (YRI) European (EUR) Utah residents with European ancestry (CEU) 94. Finnish in Finland (FIN) 93. British in England and Scotland (GBR) Iberian populations in Spain (IBS) Toscani in Italy (TSI) East Asian (EAS) Chinese Dai in Xishuangbanna, China (CDX) Han Chinese in Bejing, China (CHB) 103. Southern Han Chinese, China (CHS) 105. Japanese in Tokyo, Japan (JPT) Kinh in Ho Chi Minh City, Vietnam (KHV) South Asian (SAS) 489. Bengali in Bangladesh (BEB) 86. Gujarati Indian in Houston,Texas (GIH) Indian Telugu in the United Kingdom (ITU) Punjabi in Lahore, Pakistan (PJL) Sri Lankan Tamil in the United Kingdom (STU) Admixed American (ADM) Colombian in Medellin, Colombia (CLM) 60. Mexican Ancestry in Los Angeles, California (MXL) 64. Peruvian in Lima, Peru (PEL) Puerto Rican in Puerto Rico (PUR) 55. TOTAL")
20
Minimiser le LD entre les SNPs
FSuite Minimiser le LD entre les SNPs Géno HBD Sélectionner une sous-carte minimise le LD entre les SNPs, mais entraîne une perte d'information FSuite_HOTS : 100 sous-cartes aléatoires délimitées par les points chauds de recombinaison. Contrôle qualité: seules les sous-cartes où a<1 sont gardées => Q-Score : nombre de cartes valides par individu

21
Points chauds de recombinaison (McVean et coll. 2004, Winckler et coll
YRI CEU JPT-CHB 40 30 20 10 - points chauds de recombinaison ayant une intensité >10cM/Mb

22
RELPAIR (Epstein et al. 2000)
Dépend des fréquences alléliques Géno IBD Les probabilités de transition dépendent de la relation étudiée RELPAIR calcule la vraisemblance de 8 relations d’apparentement Ne modélise pas le LD… (application de la méthode des sous cartes) MZ -- MONOZYGOTIC TWINS FULL(FS) -- FULL SIBS P/OFF(PO) -- PARENT/OFFSPRING HALF(HS) -- HALF SIBS GP/GC(GG) -- GRANDPARENT/GRANDCHILD AVNC(AV) -- AVUNCULAR COUS(CO) -- FIRST COUSINS UNREL(UN) – UNRELATED
 MZ -- MONOZYGOTIC TWINS FULL(FS) -- FULL SIBS. P/OFF(PO) -- PARENT/OFFSPRING HALF(HS) -- HALF SIBS. GP/GC(GG) -- GRANDPARENT/GRANDCHILD AVNC(AV) -- AVUNCULAR. COUS(CO) -- FIRST COUSINS UNREL(UN) – UNRELATED.")
23
Simulations nous avons simulé des échantillons dans lesquels les individus avaient des proportions différentes de génomes d’origine européenne CEU et africaine YRI. 100 échantillons de 300 individus avec un niveau de consanguinité différent 6 (1C), 6 (2C), 18 (3C), 30 (4C), 240 (OUT) Pour avoir un modèle réaliste du LD nous avons utilisé les haplotypes du panel HapMap 3 232 haplotypes européens (CEU) et 226 d’origine africaines (YRI); typés sur la puce 987k 4 fréquences alléliques différentes sont appliquées : CEU, YRI, JPT+CHB et de l’échantillon Nous comparons ainsi les méthodes simple-point (PLINK) et multipoints (FSuite) avec tous ces paramètres vis-à-vis du facteur de consanguinité f Δf = festimé - fvrai
, 6 (2C), 18 (3C), 30 (4C), 240 (OUT) Pour avoir un modèle réaliste du LD nous avons utilisé les haplotypes du panel. HapMap haplotypes européens (CEU) et 226 d’origine africaines (YRI); typés sur la puce 987k. 4 fréquences alléliques différentes sont appliquées : CEU, YRI, JPT+CHB et de l’échantillon. Nous comparons ainsi les méthodes simple-point (PLINK) et multipoints (FSuite) avec tous ces paramètres vis-à-vis du facteur de consanguinité f. Δf = festimé - fvrai.")
24
L’individu le plus consanguin détecté dans le panel 1000 Génomes
HG04070 : ITU indien télougou en UK avec f = 0,169 issu d’une union compatible 2x1C

Présentations similaires



 r =>")



>")












